Xiru Wu, Lingzhi Wang, Yuan Qin, Yalei Gao, Min Yang, Pei Cao, Kai Liu
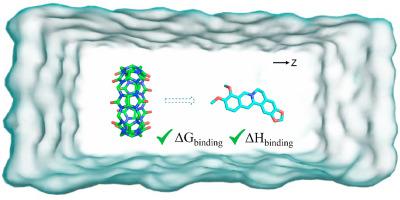
{"title":"Prediction of binding affinity and enthalpy of CB7 with alkaloids by attach-pull-release molecular dynamics simulations study","authors":"Xiru Wu, Lingzhi Wang, Yuan Qin, Yalei Gao, Min Yang, Pei Cao, Kai Liu","doi":"10.1016/j.jmgm.2024.108810","DOIUrl":null,"url":null,"abstract":"<div><p>Host-guest complex has attracted much attention because of their fantastic capability. Accurate prediction of their binding affinity and enthalpy is essential to the rational design of guest molecules. The attach-pull-release (APR) method proposed by Henriksen et al. (J. Chem. Theory Comput., 2015, 11:4377.) shows good prediction capability of binding affinity especially for host-guest system. In order to further evaluate the performance of APR method in practice, we have conducted the calculations on the macrocycle cucurbit [7]urils (CB7) encapsulated with four structurally similar alkaloids (berberine, coptisine, epiberberine and palmatine) with two force fields (GAFF and GAFF2) and three water models (TIP3P, SPC/E and OPC). Compared to the experimental data, the calculation by the combination of GAFF2 and SPC/E force field presents the best performance, of which the Pearson correlation coefficients (R<sup>2</sup>) is 0.95, and the root-mean-square-deviation is 3.04 kcal/mol. While the predictions from GAFF force field all overestimated the binding affinity, suggesting a systematic error may be involved. Comparison of calculation also indicates that the accuracy of prediction was susceptible to the combination of force field. Therefore, it would be necessary to repeat the simulation with different combination of force fields in practice.</p></div>","PeriodicalId":16361,"journal":{"name":"Journal of molecular graphics & modelling","volume":"131 ","pages":"Article 108810"},"PeriodicalIF":3.0000,"publicationDate":"2024-06-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular graphics & modelling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1093326324001104","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Host-guest complex has attracted much attention because of their fantastic capability. Accurate prediction of their binding affinity and enthalpy is essential to the rational design of guest molecules. The attach-pull-release (APR) method proposed by Henriksen et al. (J. Chem. Theory Comput., 2015, 11:4377.) shows good prediction capability of binding affinity especially for host-guest system. In order to further evaluate the performance of APR method in practice, we have conducted the calculations on the macrocycle cucurbit [7]urils (CB7) encapsulated with four structurally similar alkaloids (berberine, coptisine, epiberberine and palmatine) with two force fields (GAFF and GAFF2) and three water models (TIP3P, SPC/E and OPC). Compared to the experimental data, the calculation by the combination of GAFF2 and SPC/E force field presents the best performance, of which the Pearson correlation coefficients (R2) is 0.95, and the root-mean-square-deviation is 3.04 kcal/mol. While the predictions from GAFF force field all overestimated the binding affinity, suggesting a systematic error may be involved. Comparison of calculation also indicates that the accuracy of prediction was susceptible to the combination of force field. Therefore, it would be necessary to repeat the simulation with different combination of force fields in practice.
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
