Chen-Chen Er, Cheng-May Fung, Wei-Kean Chong, Yong Jieh Lee, Lling-Lling Tan, Yee Sin Ang, Nikhil V. Medhekar, Siang-Piao Chai
{"title":"Computational Design of 2D Phosphorus Nanostructures for Renewable Energy Applications: A Review","authors":"Chen-Chen Er, Cheng-May Fung, Wei-Kean Chong, Yong Jieh Lee, Lling-Lling Tan, Yee Sin Ang, Nikhil V. Medhekar, Siang-Piao Chai","doi":"10.1002/aelm.202300869","DOIUrl":null,"url":null,"abstract":"<p>Elemental phosphorus in its various allotropes has received tremendous research attention recently due to its intriguing electronic and structural properties. Notably, the application of nanostructured materials to overcome the inherent flaws in bulk materials is promising. However, many challenges need to be addressed before its widespread implementation. Thus, a specific tenet to design novel and robust nanomaterials is a decisive factor in the desired outcome, and the most daunting task before realizing this is solving the Schrödinger equation. First principle density functional theory (DFT) calculations have emerged as an insightful and accurate design tool to investigate the structural, electronic, and possible synthesis scenarios of yet undiscovered materials at atomic levels. In this review, the basic principles and the importance of DFT are discussed, followed by a summary of recent advances in the first principle study of elemental phosphorus-based nanomaterials. Elemental phosphorus-based nanomaterials and their allotropes have attracted growing interest in the renewable energy community due to their modulable product selectivity. However, the understanding of the physical phenomena of allotropic modification is still lacking. Therefore, the aim is to motivate experimental researchers to conduct DFT studies and experiments to comprehend relevant engineered nanomaterials better. Finally, the challenges and potential future research directions for further theoretical and computational development of phosphorus-based nanomaterials are outlined.</p>","PeriodicalId":110,"journal":{"name":"Advanced Electronic Materials","volume":"10 7","pages":""},"PeriodicalIF":5.3000,"publicationDate":"2024-06-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/aelm.202300869","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Advanced Electronic Materials","FirstCategoryId":"88","ListUrlMain":"https://advanced.onlinelibrary.wiley.com/doi/10.1002/aelm.202300869","RegionNum":2,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MATERIALS SCIENCE, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
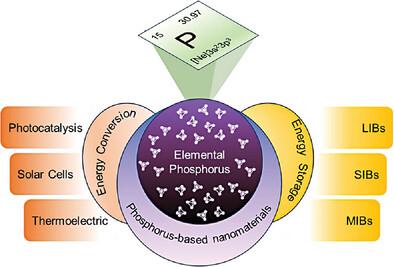
Elemental phosphorus in its various allotropes has received tremendous research attention recently due to its intriguing electronic and structural properties. Notably, the application of nanostructured materials to overcome the inherent flaws in bulk materials is promising. However, many challenges need to be addressed before its widespread implementation. Thus, a specific tenet to design novel and robust nanomaterials is a decisive factor in the desired outcome, and the most daunting task before realizing this is solving the Schrödinger equation. First principle density functional theory (DFT) calculations have emerged as an insightful and accurate design tool to investigate the structural, electronic, and possible synthesis scenarios of yet undiscovered materials at atomic levels. In this review, the basic principles and the importance of DFT are discussed, followed by a summary of recent advances in the first principle study of elemental phosphorus-based nanomaterials. Elemental phosphorus-based nanomaterials and their allotropes have attracted growing interest in the renewable energy community due to their modulable product selectivity. However, the understanding of the physical phenomena of allotropic modification is still lacking. Therefore, the aim is to motivate experimental researchers to conduct DFT studies and experiments to comprehend relevant engineered nanomaterials better. Finally, the challenges and potential future research directions for further theoretical and computational development of phosphorus-based nanomaterials are outlined.
期刊介绍:
Advanced Electronic Materials is an interdisciplinary forum for peer-reviewed, high-quality, high-impact research in the fields of materials science, physics, and engineering of electronic and magnetic materials. It includes research on physics and physical properties of electronic and magnetic materials, spintronics, electronics, device physics and engineering, micro- and nano-electromechanical systems, and organic electronics, in addition to fundamental research.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
