Syeda Sumayya Tariq , Komal Zia , Mohammad Nur-e-Alam , Dmitry Nerukh , Vladimir S. Farafonov , Zaheer Ul-Haq
{"title":"Impact of mutations in SARS-CoV-2 recombinant sub-variant XBB.1.16 on the binding affinity with human ACE2 receptor","authors":"Syeda Sumayya Tariq , Komal Zia , Mohammad Nur-e-Alam , Dmitry Nerukh , Vladimir S. Farafonov , Zaheer Ul-Haq","doi":"10.1016/j.jmgm.2024.108813","DOIUrl":null,"url":null,"abstract":"<div><p>Despite the waning threat of the COVID-19 pandemic, its detrimental impact on global health persists. Regardless of natural immunity or immunity obtained through vaccination, emerging variants of the virus continue to undergo mutations and propagate globally. The persistent mutations in SARS-CoV-2, along with the subsequent formation of recombinant sub-variants has become a challenge for researchers and health professionals, raising concerns about the efficacy of current vaccines. Gaining a better understanding of the biochemical interactions between the Spike Protein (RBD) of SARS-CoV-2 variants and the human ACE2 receptor can prove to be beneficial in designing and developing antiviral therapeutics that are equally effective against all strains and emerging variants. Our objective in this study was to investigate the interfacial binding pattern of the SARS-CoV-2 RBD-ACE2 complex of the Wild Type (WT), Omicron, and the Omicron recombinant sub-variant XBB.1.16. We aimed to examine the atomic level factors and observe how mutations influence the interaction between the virus and its host using Molecular Dynamics simulation, MM/GBSA energy calculations, and Principal Component Analysis. Our findings reveal a higher degree of structural deviation and flexibility in XBB.1.16 compared to WT and Omicron. PCA indicated a wider cluster and significant flexibility in the movements of XBB.1.16 which can also be observed in free energy landscapes, while the normal mode analysis revealed converging motions within the RBD-ACE2 complexes which can facilitate the interaction between them. A pattern of decreased binding affinity was observed in case of XBB.1.16 when compared to the WT and Omicron. These observed deviations in XBB.1.16 when compared to its parent lineage Omicron, and WT can be attributed to the mutations specific to it. Collectively, these results enhance our understanding of the impact of mutations on the interaction between this strain and the host, taking us one step closer to designing effective antiviral therapeutics against the continually mutating strains.</p></div>","PeriodicalId":16361,"journal":{"name":"Journal of molecular graphics & modelling","volume":"131 ","pages":"Article 108813"},"PeriodicalIF":3.0000,"publicationDate":"2024-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular graphics & modelling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S109332632400113X","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/6/13 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
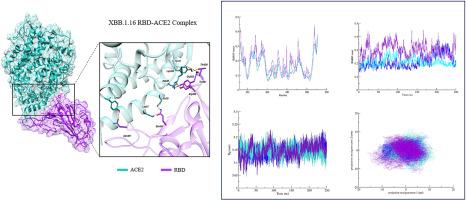
Despite the waning threat of the COVID-19 pandemic, its detrimental impact on global health persists. Regardless of natural immunity or immunity obtained through vaccination, emerging variants of the virus continue to undergo mutations and propagate globally. The persistent mutations in SARS-CoV-2, along with the subsequent formation of recombinant sub-variants has become a challenge for researchers and health professionals, raising concerns about the efficacy of current vaccines. Gaining a better understanding of the biochemical interactions between the Spike Protein (RBD) of SARS-CoV-2 variants and the human ACE2 receptor can prove to be beneficial in designing and developing antiviral therapeutics that are equally effective against all strains and emerging variants. Our objective in this study was to investigate the interfacial binding pattern of the SARS-CoV-2 RBD-ACE2 complex of the Wild Type (WT), Omicron, and the Omicron recombinant sub-variant XBB.1.16. We aimed to examine the atomic level factors and observe how mutations influence the interaction between the virus and its host using Molecular Dynamics simulation, MM/GBSA energy calculations, and Principal Component Analysis. Our findings reveal a higher degree of structural deviation and flexibility in XBB.1.16 compared to WT and Omicron. PCA indicated a wider cluster and significant flexibility in the movements of XBB.1.16 which can also be observed in free energy landscapes, while the normal mode analysis revealed converging motions within the RBD-ACE2 complexes which can facilitate the interaction between them. A pattern of decreased binding affinity was observed in case of XBB.1.16 when compared to the WT and Omicron. These observed deviations in XBB.1.16 when compared to its parent lineage Omicron, and WT can be attributed to the mutations specific to it. Collectively, these results enhance our understanding of the impact of mutations on the interaction between this strain and the host, taking us one step closer to designing effective antiviral therapeutics against the continually mutating strains.
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
