David F. Hahn*, Vytautas Gapsys, Bert L. de Groot, David L. Mobley and Gary Tresadern,
{"title":"Current State of Open Source Force Fields in Protein–Ligand Binding Affinity Predictions","authors":"David F. Hahn*, Vytautas Gapsys, Bert L. de Groot, David L. Mobley and Gary Tresadern, ","doi":"10.1021/acs.jcim.4c00417","DOIUrl":null,"url":null,"abstract":"<p >In drug discovery, the in silico prediction of binding affinity is one of the major means to prioritize compounds for synthesis. Alchemical relative binding free energy (RBFE) calculations based on molecular dynamics (MD) simulations are nowadays a popular approach for the accurate affinity ranking of compounds. MD simulations rely on empirical force field parameters, which strongly influence the accuracy of the predicted affinities. Here, we evaluate the ability of six different small-molecule force fields to predict experimental protein–ligand binding affinities in RBFE calculations on a set of 598 ligands and 22 protein targets. The public force fields OpenFF Parsley and Sage, GAFF, and CGenFF show comparable accuracy, while OPLS3e is significantly more accurate. However, a consensus approach using Sage, GAFF, and CGenFF leads to accuracy comparable to OPLS3e. While Parsley and Sage are performing comparably based on aggregated statistics across the whole dataset, there are differences in terms of outliers. Analysis of the force field reveals that improved parameters lead to significant improvement in the accuracy of affinity predictions on subsets of the dataset involving those parameters. Lower accuracy can not only be attributed to the force field parameters but is also dependent on input preparation and sampling convergence of the calculations. Especially large perturbations and nonconverged simulations lead to less accurate predictions. The input structures, Gromacs force field files, as well as the analysis Python notebooks are available on GitHub.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"64 13","pages":"5063–5076"},"PeriodicalIF":5.3000,"publicationDate":"2024-06-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/epdf/10.1021/acs.jcim.4c00417","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.4c00417","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
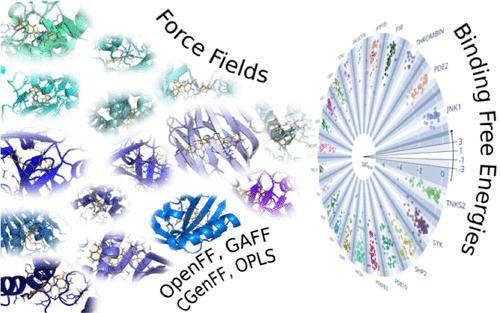
In drug discovery, the in silico prediction of binding affinity is one of the major means to prioritize compounds for synthesis. Alchemical relative binding free energy (RBFE) calculations based on molecular dynamics (MD) simulations are nowadays a popular approach for the accurate affinity ranking of compounds. MD simulations rely on empirical force field parameters, which strongly influence the accuracy of the predicted affinities. Here, we evaluate the ability of six different small-molecule force fields to predict experimental protein–ligand binding affinities in RBFE calculations on a set of 598 ligands and 22 protein targets. The public force fields OpenFF Parsley and Sage, GAFF, and CGenFF show comparable accuracy, while OPLS3e is significantly more accurate. However, a consensus approach using Sage, GAFF, and CGenFF leads to accuracy comparable to OPLS3e. While Parsley and Sage are performing comparably based on aggregated statistics across the whole dataset, there are differences in terms of outliers. Analysis of the force field reveals that improved parameters lead to significant improvement in the accuracy of affinity predictions on subsets of the dataset involving those parameters. Lower accuracy can not only be attributed to the force field parameters but is also dependent on input preparation and sampling convergence of the calculations. Especially large perturbations and nonconverged simulations lead to less accurate predictions. The input structures, Gromacs force field files, as well as the analysis Python notebooks are available on GitHub.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
