Walter Filgueira de Azevedo Jr, Rodrigo Quiroga, Marcos Ariel Villarreal, Nelson José Freitas da Silveira, Gabriela Bitencourt-Ferreira, Amauri Duarte da Silva, Martina Veit-Acosta, Patricia Rufino Oliveira, Marco Tutone, Nadezhda Biziukova, Vladimir Poroikov, Olga Tarasova, Stéphaine Baud
{"title":"SAnDReS 2.0: Development of machine-learning models to explore the scoring function space","authors":"Walter Filgueira de Azevedo Jr, Rodrigo Quiroga, Marcos Ariel Villarreal, Nelson José Freitas da Silveira, Gabriela Bitencourt-Ferreira, Amauri Duarte da Silva, Martina Veit-Acosta, Patricia Rufino Oliveira, Marco Tutone, Nadezhda Biziukova, Vladimir Poroikov, Olga Tarasova, Stéphaine Baud","doi":"10.1002/jcc.27449","DOIUrl":null,"url":null,"abstract":"<p>Classical scoring functions may exhibit low accuracy in determining ligand binding affinity for proteins. The availability of both protein–ligand structures and affinity data make it possible to develop machine-learning models focused on specific protein systems with superior predictive performance. Here, we report a new methodology named SAnDReS that combines AutoDock Vina 1.2 with 54 regression methods available in Scikit-Learn to calculate binding affinity based on protein–ligand structures. This approach allows exploration of the scoring function space. SAnDReS generates machine-learning models based on crystal, docked, and AlphaFold-generated structures. As a proof of concept, we examine the performance of SAnDReS-generated models in three case studies. For all three cases, our models outperformed classical scoring functions. Also, SAnDReS-generated models showed predictive performance close to or better than other machine-learning models such as <i>K</i><sub>DEEP</sub>, CSM-lig, and Δ<sub>Vina</sub>RF<sub>20</sub>. SAnDReS 2.0 is available to download at https://github.com/azevedolab/sandres.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 27","pages":"2333-2346"},"PeriodicalIF":4.8000,"publicationDate":"2024-06-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27449","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
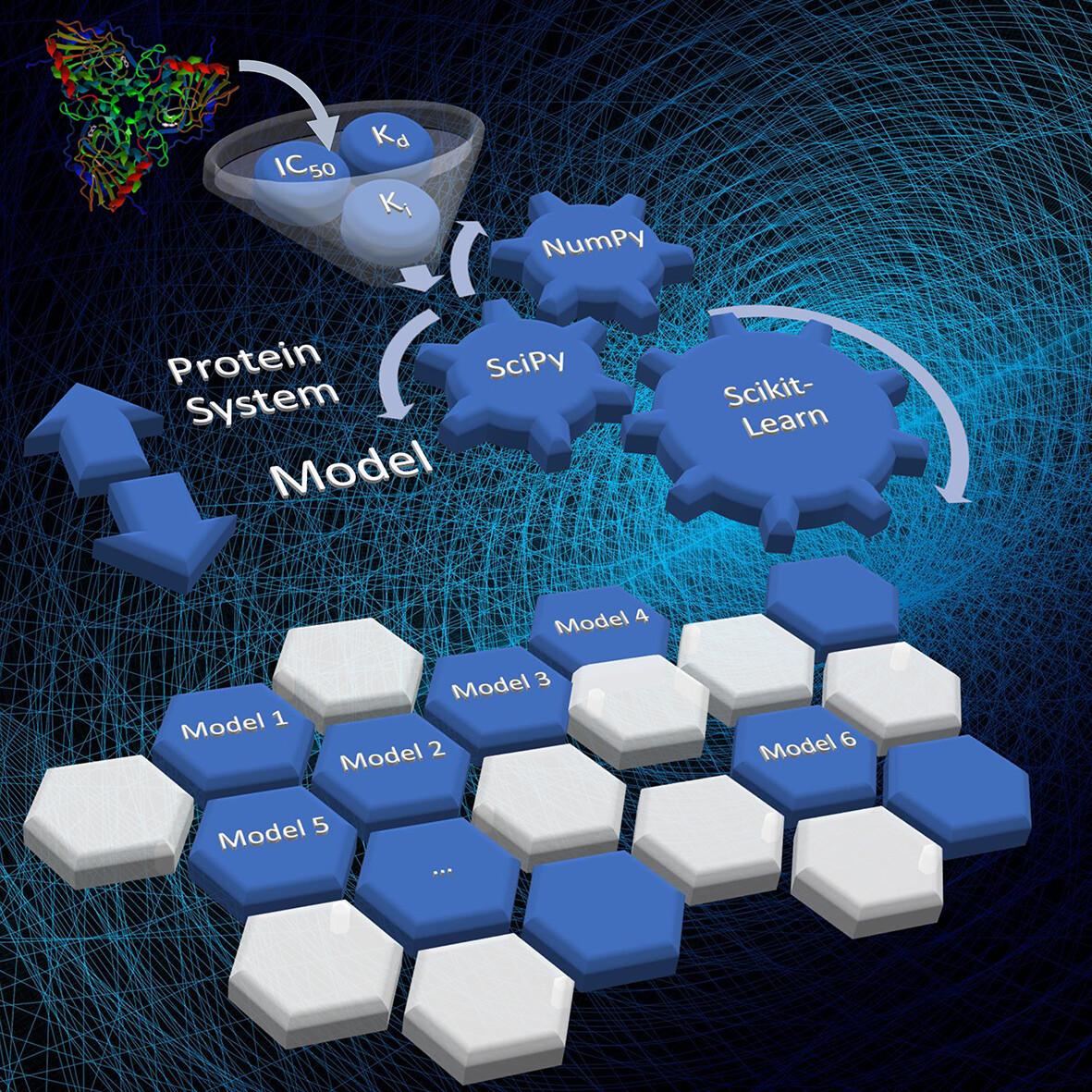
Classical scoring functions may exhibit low accuracy in determining ligand binding affinity for proteins. The availability of both protein–ligand structures and affinity data make it possible to develop machine-learning models focused on specific protein systems with superior predictive performance. Here, we report a new methodology named SAnDReS that combines AutoDock Vina 1.2 with 54 regression methods available in Scikit-Learn to calculate binding affinity based on protein–ligand structures. This approach allows exploration of the scoring function space. SAnDReS generates machine-learning models based on crystal, docked, and AlphaFold-generated structures. As a proof of concept, we examine the performance of SAnDReS-generated models in three case studies. For all three cases, our models outperformed classical scoring functions. Also, SAnDReS-generated models showed predictive performance close to or better than other machine-learning models such as KDEEP, CSM-lig, and ΔVinaRF20. SAnDReS 2.0 is available to download at https://github.com/azevedolab/sandres.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
