{"title":"A novel mutation in SORD gene associated with distal hereditary motor neuropathies.","authors":"Xiaoqin Yuan, Shanshan Zhang, Huifang Shang, Yufeng Tang","doi":"10.1186/s12920-024-01940-5","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Distal hereditary motor neuropathy (dHMN) is a heterogeneous group of hereditary diseases caused by the gradual degeneration of the lower motor neuron. More than 30 genes associated with dHMN have been reported, while 70-80% of those with the condition are still unable to receive a genetic diagnosis.</p><p><strong>Methods: </strong>A 26-year-old man experiencing gradual weakness in his lower limbs was referred to our hospital, and data on clinical features, laboratory tests, and electrophysiological tests were collected. To identify the disease-causing mutation, we conducted whole exome sequencing (WES) and then validated it through Sanger sequencing for the proband and his parents. Silico analysis was performed to predict the pathogenesis of the identified mutations. A literature review of all reported mutations of the related gene for the disease was performed.</p><p><strong>Results: </strong>The patient presented with dHMN phenotype harboring a novel homozygous variant c.361G > C (p.Ala121Pro) in SORD, inherited from his parents, respectively. A121 is a highly conserved site and the mutation was categorized as \"likely pathogenic\" according to the criteria and guidelines of the American College of Medical Genetics and Genomics (ACMG). A total of 13 published articles including 101 patients reported 18 SORD variants. Almost all described cases have the homozygous deletion variant c.757delG (p.A253Qfs*27) or compound heterozygous state of a combination of c.757delG (p.A253Qfs*27) with another variant. The variant c.361G > C (p.Ala121Pro) detected in our patient was the second homozygous variant in SORD-associated hereditary neuropathy.</p><p><strong>Conclusion: </strong>One novel homozygous variant c.361G > C (p.Ala121Pro) in SORD was identified in a Chinese patient with dHMN phenotype, which expands the mutation spectrum of SORD-associated hereditary neuropathy and underscores the significance of screening for SORD variants in patients with undiagnosed hereditary neuropathy patients.</p>","PeriodicalId":8915,"journal":{"name":"BMC Medical Genomics","volume":"17 1","pages":"169"},"PeriodicalIF":2.0000,"publicationDate":"2024-06-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11194961/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Medical Genomics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12920-024-01940-5","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Distal hereditary motor neuropathy (dHMN) is a heterogeneous group of hereditary diseases caused by the gradual degeneration of the lower motor neuron. More than 30 genes associated with dHMN have been reported, while 70-80% of those with the condition are still unable to receive a genetic diagnosis.
Methods: A 26-year-old man experiencing gradual weakness in his lower limbs was referred to our hospital, and data on clinical features, laboratory tests, and electrophysiological tests were collected. To identify the disease-causing mutation, we conducted whole exome sequencing (WES) and then validated it through Sanger sequencing for the proband and his parents. Silico analysis was performed to predict the pathogenesis of the identified mutations. A literature review of all reported mutations of the related gene for the disease was performed.
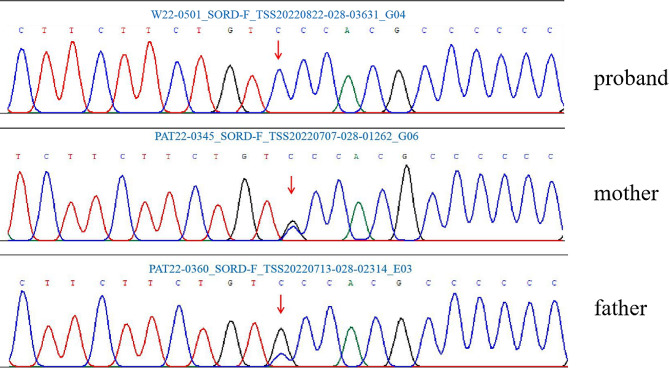
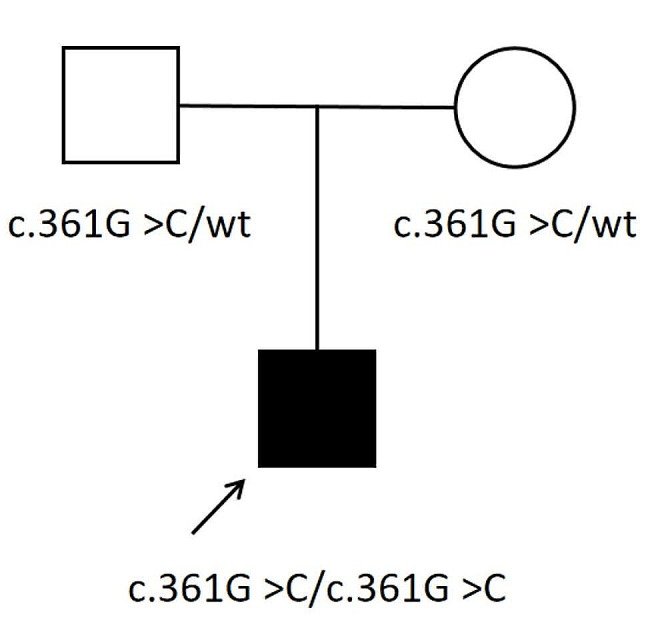
Results: The patient presented with dHMN phenotype harboring a novel homozygous variant c.361G > C (p.Ala121Pro) in SORD, inherited from his parents, respectively. A121 is a highly conserved site and the mutation was categorized as "likely pathogenic" according to the criteria and guidelines of the American College of Medical Genetics and Genomics (ACMG). A total of 13 published articles including 101 patients reported 18 SORD variants. Almost all described cases have the homozygous deletion variant c.757delG (p.A253Qfs*27) or compound heterozygous state of a combination of c.757delG (p.A253Qfs*27) with another variant. The variant c.361G > C (p.Ala121Pro) detected in our patient was the second homozygous variant in SORD-associated hereditary neuropathy.
Conclusion: One novel homozygous variant c.361G > C (p.Ala121Pro) in SORD was identified in a Chinese patient with dHMN phenotype, which expands the mutation spectrum of SORD-associated hereditary neuropathy and underscores the significance of screening for SORD variants in patients with undiagnosed hereditary neuropathy patients.
期刊介绍:
BMC Medical Genomics is an open access journal publishing original peer-reviewed research articles in all aspects of functional genomics, genome structure, genome-scale population genetics, epigenomics, proteomics, systems analysis, and pharmacogenomics in relation to human health and disease.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
