Impact of Nonnative Interactions on the Binding Kinetics of Intrinsically Disordered p53 with MDM2: Insights from All-Atom Simulation and Markov State Model Analysis
Qianjun Xu, Maohua Yang, Jie Ji, Jingwei Weng*, Wenning Wang* and Xin Xu*,
{"title":"Impact of Nonnative Interactions on the Binding Kinetics of Intrinsically Disordered p53 with MDM2: Insights from All-Atom Simulation and Markov State Model Analysis","authors":"Qianjun Xu, Maohua Yang, Jie Ji, Jingwei Weng*, Wenning Wang* and Xin Xu*, ","doi":"10.1021/acs.jcim.3c01833","DOIUrl":null,"url":null,"abstract":"<p >Intrinsically disordered proteins (IDPs) lack a well-defined tertiary structure but are essential players in various biological processes. Their ability to undergo a disorder-to-order transition upon binding to their partners, known as the folding-upon-binding process, is crucial for their function. One classical example is the intrinsically disordered transactivation domain (TAD) of the tumor suppressor protein p53, which quickly forms a structured α-helix after binding to its partner MDM2, with clinical significance for cancer treatment. However, the contribution of nonnative interactions between the IDP and its partner to the rapid binding kinetics, as well as their interplay with native interactions, is not well understood at the atomic level. Here, we used molecular dynamics simulation and Markov state model (MSM) analysis to study the folding-upon-binding mechanism between p53-TAD and MDM2. Our results suggest that the system progresses from the nascent encounter complex to the well-structured encounter complex and finally reaches the native complex, following an induced-fit mechanism. We found that nonnative hydrophobic and hydrogen bond interactions, combined with native interactions, effectively stabilize the nascent and well-structured encounter complexes. Among the nonnative interactions, Leu25<sup>p53</sup>–Leu54<sup>MDM2</sup> and Leu25<sup>p53</sup>–Phe55<sup>MDM2</sup> are particularly noteworthy, as their interaction strength is close to the optimum. Evidently, strengthening or weakening these interactions could both adversely affect the binding kinetics. Overall, our findings suggest that nonnative interactions are evolutionarily optimized to accelerate the binding kinetics of IDPs in conjunction with native interactions.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"64 13","pages":"5219–5231"},"PeriodicalIF":5.3000,"publicationDate":"2024-06-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.3c01833","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
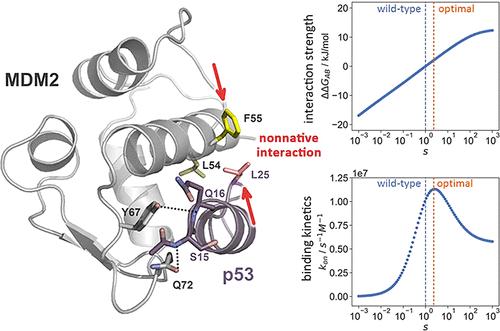
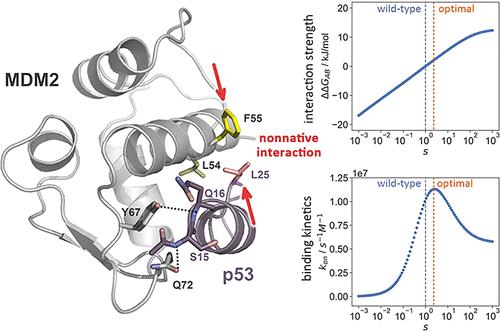
Intrinsically disordered proteins (IDPs) lack a well-defined tertiary structure but are essential players in various biological processes. Their ability to undergo a disorder-to-order transition upon binding to their partners, known as the folding-upon-binding process, is crucial for their function. One classical example is the intrinsically disordered transactivation domain (TAD) of the tumor suppressor protein p53, which quickly forms a structured α-helix after binding to its partner MDM2, with clinical significance for cancer treatment. However, the contribution of nonnative interactions between the IDP and its partner to the rapid binding kinetics, as well as their interplay with native interactions, is not well understood at the atomic level. Here, we used molecular dynamics simulation and Markov state model (MSM) analysis to study the folding-upon-binding mechanism between p53-TAD and MDM2. Our results suggest that the system progresses from the nascent encounter complex to the well-structured encounter complex and finally reaches the native complex, following an induced-fit mechanism. We found that nonnative hydrophobic and hydrogen bond interactions, combined with native interactions, effectively stabilize the nascent and well-structured encounter complexes. Among the nonnative interactions, Leu25p53–Leu54MDM2 and Leu25p53–Phe55MDM2 are particularly noteworthy, as their interaction strength is close to the optimum. Evidently, strengthening or weakening these interactions could both adversely affect the binding kinetics. Overall, our findings suggest that nonnative interactions are evolutionarily optimized to accelerate the binding kinetics of IDPs in conjunction with native interactions.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
