{"title":"TransfIGN: A Structure-Based Deep Learning Method for Modeling the Interaction between HLA-A*02:01 and Antigen Peptides","authors":"Nanqi Hong, Dejun Jiang, Zhe Wang, Huiyong Sun, Hao Luo, Lingjie Bao, Mingli Song*, Yu Kang* and Tingjun Hou*, ","doi":"10.1021/acs.jcim.4c00678","DOIUrl":null,"url":null,"abstract":"<p >The intricate interaction between major histocompatibility complexes (MHCs) and antigen peptides with diverse amino acid sequences plays a pivotal role in immune responses and T cell activity. In recent years, deep learning (DL)-based models have emerged as promising tools for accelerating antigen peptide screening. However, most of these models solely rely on one-dimensional amino acid sequences, overlooking crucial information required for the three-dimensional (3-D) space binding process. In this study, we propose TransfIGN, a structure-based DL model that is inspired by our previously developed framework, Interaction Graph Network (IGN), and incorporates sequence information from transformers to predict the interactions between HLA-A*02:01 and antigen peptides. Our model, trained on a comprehensive data set containing 61,816 sequences with 9051 binding affinity labels and 56,848 eluted ligand labels, achieves an area under the curve (AUC) of 0.893 on the binary data set, better than state-of-the-art sequence-based models trained on larger data sets such as NetMHCpan4.1, ANN, and TransPHLA. Furthermore, when evaluated on the IEDB weekly benchmark data sets, our predictions (AUC = 0.816) are better than those of the recommended methods like the IEDB consensus (AUC = 0.795). Notably, the interaction weight matrices generated by our method highlight the strong interactions at specific positions within peptides, emphasizing the model’s ability to provide physical interpretability. This capability to unveil binding mechanisms through intricate structural features holds promise for new immunotherapeutic avenues.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"64 13","pages":"5016–5027"},"PeriodicalIF":5.3000,"publicationDate":"2024-06-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.4c00678","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
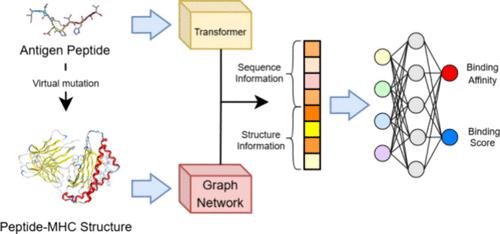
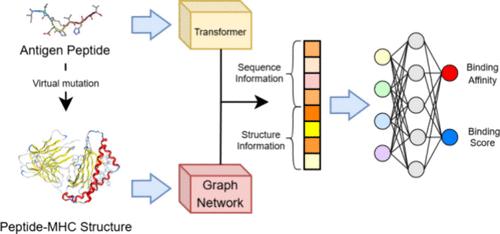
The intricate interaction between major histocompatibility complexes (MHCs) and antigen peptides with diverse amino acid sequences plays a pivotal role in immune responses and T cell activity. In recent years, deep learning (DL)-based models have emerged as promising tools for accelerating antigen peptide screening. However, most of these models solely rely on one-dimensional amino acid sequences, overlooking crucial information required for the three-dimensional (3-D) space binding process. In this study, we propose TransfIGN, a structure-based DL model that is inspired by our previously developed framework, Interaction Graph Network (IGN), and incorporates sequence information from transformers to predict the interactions between HLA-A*02:01 and antigen peptides. Our model, trained on a comprehensive data set containing 61,816 sequences with 9051 binding affinity labels and 56,848 eluted ligand labels, achieves an area under the curve (AUC) of 0.893 on the binary data set, better than state-of-the-art sequence-based models trained on larger data sets such as NetMHCpan4.1, ANN, and TransPHLA. Furthermore, when evaluated on the IEDB weekly benchmark data sets, our predictions (AUC = 0.816) are better than those of the recommended methods like the IEDB consensus (AUC = 0.795). Notably, the interaction weight matrices generated by our method highlight the strong interactions at specific positions within peptides, emphasizing the model’s ability to provide physical interpretability. This capability to unveil binding mechanisms through intricate structural features holds promise for new immunotherapeutic avenues.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
