Neus Pagès-Vilà , Ilaria Gamba , Martin Clémancey , Jean-Marc Latour , Anna Company , Miquel Costas
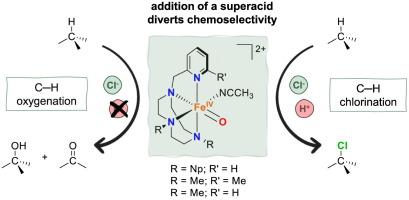
{"title":"Proton-triggered chemoselective halogenation of aliphatic C–H bonds with nonheme FeIV-oxo complexes","authors":"Neus Pagès-Vilà , Ilaria Gamba , Martin Clémancey , Jean-Marc Latour , Anna Company , Miquel Costas","doi":"10.1016/j.jinorgbio.2024.112643","DOIUrl":null,"url":null,"abstract":"<div><p>Halogenation of aliphatic C–H bonds is a chemical transformation performed in nature by mononuclear nonheme iron dependent halogenases. The mechanism involves the formation of an iron(IV)-oxo-chloride species that abstracts the hydrogen atom from the reactive C–H bond to form a carbon-centered radical that selectively reacts with the bound chloride ligand, a process commonly referred to as halide rebound. The factors that determine the halide rebound, as opposed to the reaction with the incipient hydroxide ligand, are not clearly understood and examples of well-defined iron(IV)-oxo-halide compounds competent in C–H halogenation are scarce. In this work we have studied the reactivity of three well-defined iron(IV)-oxo complexes containing variants of the tetradentate 1-(2-pyridylmethyl)-1,4,7-triazacyclononane ligand (Pytacn). Interestingly, these compounds exhibit a change in their chemoselectivity towards the functionalization of C–H bonds under certain conditions: their reaction towards C–H bonds in the presence of a halide anionleads to exclusive oxygenation, while the addition of a superacid results in halogenation. Almost quantitative halogenation of ethylbenzene is observed when using the two systems with more sterically congested ligands and even the chlorination of strong C–H bonds such as those of cyclohexane is performed when a methyl group is present in the sixth position of the pyridine ring of the ligand. Mechanistic studies suggest that both reactions, oxygenation and halogenation, proceed through a common rate determining hydrogen atom transfer step and the presence of the acid dictates the fate of the resulting alkyl radical towards preferential halogenation over oxygenation.</p></div>","PeriodicalId":364,"journal":{"name":"Journal of Inorganic Biochemistry","volume":"259 ","pages":"Article 112643"},"PeriodicalIF":3.1000,"publicationDate":"2024-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S0162013424001673/pdfft?md5=5626544c260c888279f2b8a1d6e1f6e1&pid=1-s2.0-S0162013424001673-main.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Inorganic Biochemistry","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0162013424001673","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/6/17 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Halogenation of aliphatic C–H bonds is a chemical transformation performed in nature by mononuclear nonheme iron dependent halogenases. The mechanism involves the formation of an iron(IV)-oxo-chloride species that abstracts the hydrogen atom from the reactive C–H bond to form a carbon-centered radical that selectively reacts with the bound chloride ligand, a process commonly referred to as halide rebound. The factors that determine the halide rebound, as opposed to the reaction with the incipient hydroxide ligand, are not clearly understood and examples of well-defined iron(IV)-oxo-halide compounds competent in C–H halogenation are scarce. In this work we have studied the reactivity of three well-defined iron(IV)-oxo complexes containing variants of the tetradentate 1-(2-pyridylmethyl)-1,4,7-triazacyclononane ligand (Pytacn). Interestingly, these compounds exhibit a change in their chemoselectivity towards the functionalization of C–H bonds under certain conditions: their reaction towards C–H bonds in the presence of a halide anionleads to exclusive oxygenation, while the addition of a superacid results in halogenation. Almost quantitative halogenation of ethylbenzene is observed when using the two systems with more sterically congested ligands and even the chlorination of strong C–H bonds such as those of cyclohexane is performed when a methyl group is present in the sixth position of the pyridine ring of the ligand. Mechanistic studies suggest that both reactions, oxygenation and halogenation, proceed through a common rate determining hydrogen atom transfer step and the presence of the acid dictates the fate of the resulting alkyl radical towards preferential halogenation over oxygenation.
期刊介绍:
The Journal of Inorganic Biochemistry is an established international forum for research in all aspects of Biological Inorganic Chemistry. Original papers of a high scientific level are published in the form of Articles (full length papers), Short Communications, Focused Reviews and Bioinorganic Methods. Topics include: the chemistry, structure and function of metalloenzymes; the interaction of inorganic ions and molecules with proteins and nucleic acids; the synthesis and properties of coordination complexes of biological interest including both structural and functional model systems; the function of metal- containing systems in the regulation of gene expression; the role of metals in medicine; the application of spectroscopic methods to determine the structure of metallobiomolecules; the preparation and characterization of metal-based biomaterials; and related systems. The emphasis of the Journal is on the structure and mechanism of action of metallobiomolecules.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
