Renato Santos, Víctor Moreno-Torres, Ilduara Pintos, Octavio Corral, Carmen de Mendoza, Vicente Soriano, Manuel Corpas
{"title":"Low-coverage whole genome sequencing for a highly selective cohort of severe COVID-19 patients.","authors":"Renato Santos, Víctor Moreno-Torres, Ilduara Pintos, Octavio Corral, Carmen de Mendoza, Vicente Soriano, Manuel Corpas","doi":"10.46471/gigabyte.127","DOIUrl":null,"url":null,"abstract":"<p><p>Despite the advances in genetic marker identification associated with severe COVID-19, the full genetic characterisation of the disease remains elusive. This study explores imputation in low-coverage whole genome sequencing for a severe COVID-19 patient cohort. We generated a dataset of 79 imputed variant call format files using the GLIMPSE1 tool, each containing an average of 9.5 million single nucleotide variants. Validation revealed a high imputation accuracy (squared Pearson correlation ≍0.97) across sequencing platforms, showcasing GLIMPSE1's ability to confidently impute variants with minor allele frequencies as low as 2% in individuals with Spanish ancestry. We carried out a comprehensive analysis of the patient cohort, examining hospitalisation and intensive care utilisation, sex and age-based differences, and clinical phenotypes using a standardised set of medical terms developed to characterise severe COVID-19 symptoms. The methods and findings presented here can be leveraged for future genomic projects to gain vital insights into health challenges like COVID-19.</p>","PeriodicalId":73157,"journal":{"name":"GigaByte (Hong Kong, China)","volume":"2024 ","pages":"gigabyte127"},"PeriodicalIF":1.2000,"publicationDate":"2024-06-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11211761/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"GigaByte (Hong Kong, China)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.46471/gigabyte.127","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
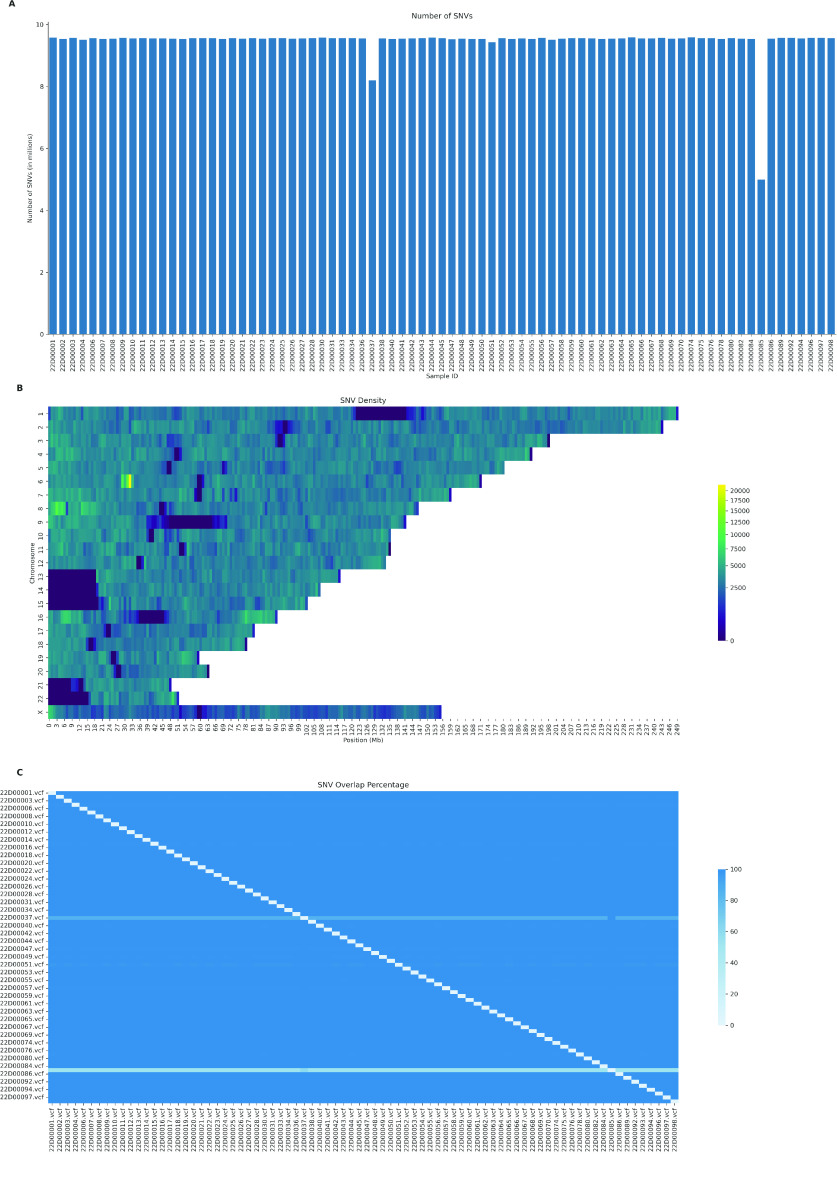
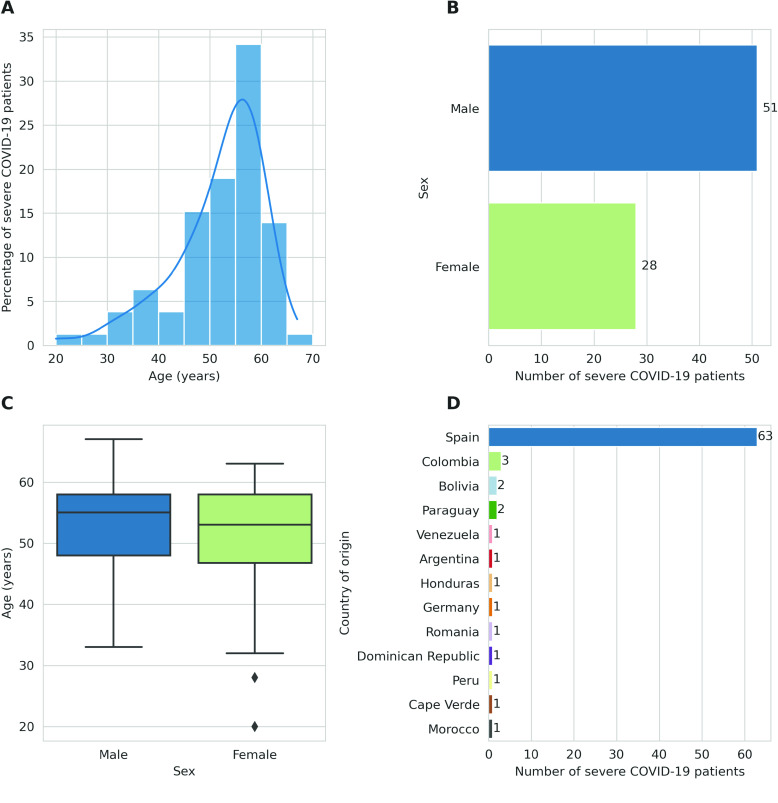
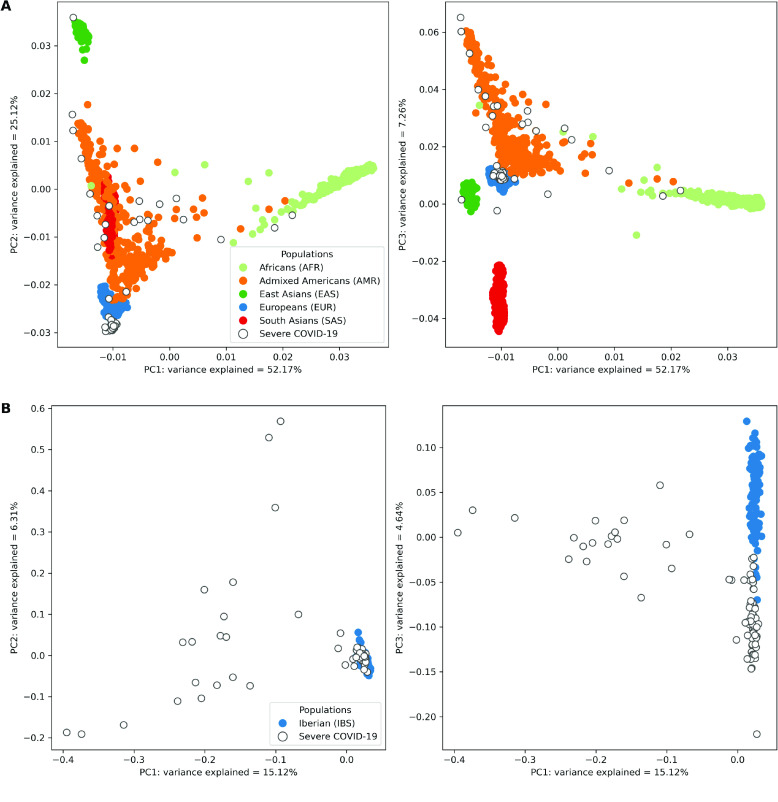
Despite the advances in genetic marker identification associated with severe COVID-19, the full genetic characterisation of the disease remains elusive. This study explores imputation in low-coverage whole genome sequencing for a severe COVID-19 patient cohort. We generated a dataset of 79 imputed variant call format files using the GLIMPSE1 tool, each containing an average of 9.5 million single nucleotide variants. Validation revealed a high imputation accuracy (squared Pearson correlation ≍0.97) across sequencing platforms, showcasing GLIMPSE1's ability to confidently impute variants with minor allele frequencies as low as 2% in individuals with Spanish ancestry. We carried out a comprehensive analysis of the patient cohort, examining hospitalisation and intensive care utilisation, sex and age-based differences, and clinical phenotypes using a standardised set of medical terms developed to characterise severe COVID-19 symptoms. The methods and findings presented here can be leveraged for future genomic projects to gain vital insights into health challenges like COVID-19.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
