Fangting Li, Kun Lv, Xiaohua Liu, Yuqiao Zhou, Kai Liu
{"title":"Accurately Computing the Interacted Volume of Molecules over Their 3D Mesh Models.","authors":"Fangting Li, Kun Lv, Xiaohua Liu, Yuqiao Zhou, Kai Liu","doi":"10.1021/acs.jcim.4c00641","DOIUrl":null,"url":null,"abstract":"<p><p>For quickly predicting the rational arrangement of catalysts and substrates, we previously proposed a method to calculate the interacted volumes of molecules over their 3D point cloud models. However, the nonuniform density in molecular point clouds may lead to incomplete contours in some slices, reducing the accuracy of the previous method. In this paper, we propose a two-step method for more accurately computing molecular interacted volumes. First, by employing a prematched mesh slicing method, we layer the 3D triangular mesh models of the electrostatic potential isosurfaces of two molecules globally, transforming the volume calculation into finding the intersecting areas in each layer. Next, by subdividing polygonal edges, we accurately identify intersecting parts within each layer, ensuring precise calculation of interacted volumes. In addition, we present a concise overview for computing intersecting areas in cases of multiple contour intersections and for improving computational efficiency by incorporating bounding boxes at three stages. Experimental results demonstrate that our method maintains high accuracy in different experimental data sets, with an average relative error of 0.16%. On the same experimental setup, our average relative error is 0.07%, which is lower than the previous algorithm's 1.73%, improving the accuracy and stability in calculating interacted volumes.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":" ","pages":"5535-5546"},"PeriodicalIF":5.3000,"publicationDate":"2024-07-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.4c00641","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/4 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
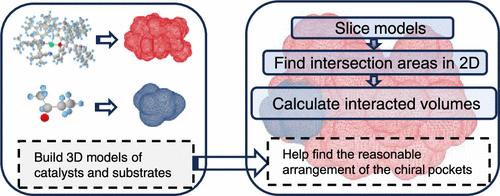
For quickly predicting the rational arrangement of catalysts and substrates, we previously proposed a method to calculate the interacted volumes of molecules over their 3D point cloud models. However, the nonuniform density in molecular point clouds may lead to incomplete contours in some slices, reducing the accuracy of the previous method. In this paper, we propose a two-step method for more accurately computing molecular interacted volumes. First, by employing a prematched mesh slicing method, we layer the 3D triangular mesh models of the electrostatic potential isosurfaces of two molecules globally, transforming the volume calculation into finding the intersecting areas in each layer. Next, by subdividing polygonal edges, we accurately identify intersecting parts within each layer, ensuring precise calculation of interacted volumes. In addition, we present a concise overview for computing intersecting areas in cases of multiple contour intersections and for improving computational efficiency by incorporating bounding boxes at three stages. Experimental results demonstrate that our method maintains high accuracy in different experimental data sets, with an average relative error of 0.16%. On the same experimental setup, our average relative error is 0.07%, which is lower than the previous algorithm's 1.73%, improving the accuracy and stability in calculating interacted volumes.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
