{"title":"Learning Force Field Parameters from Differentiable Particle-Field Molecular Dynamics.","authors":"Manuel Carrer, Henrique Musseli Cezar, Sigbjørn Løland Bore, Morten Ledum, Michele Cascella","doi":"10.1021/acs.jcim.4c00564","DOIUrl":null,"url":null,"abstract":"<p><p>We develop ∂-HylleraasMD (∂-HyMD), a fully end-to-end differentiable molecular dynamics software based on the Hamiltonian hybrid particle-field formalism, and use it to establish a protocol for automated optimization of force field parameters. ∂-HyMD is templated on the recently released HylleraaasMD software, while using the JAX autodiff framework as the main engine for the differentiable dynamics. ∂-HyMD exploits an embarrassingly parallel optimization algorithm by spawning independent simulations, whose trajectories are simultaneously processed by reverse mode automatic differentiation to calculate the gradient of the loss function, which is in turn used for iterative optimization of the force-field parameters. We show that parallel organization facilitates the convergence of the minimization procedure, avoiding the known memory and numerical stability issues of differentiable molecular dynamics approaches. We showcase the effectiveness of our implementation by producing a library of force field parameters for standard phospholipids, with either zwitterionic or anionic heads and with saturated or unsaturated tails. Compared to the all-atom reference, the force field obtained by ∂-HyMD yields better density profiles than the parameters derived from previously utilized gradient-free optimization procedures. Moreover, ∂-HyMD models can predict with good accuracy properties not included in the learning objective, such as lateral pressure profiles, and are transferable to other systems, including triglycerides.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":" ","pages":"5510-5520"},"PeriodicalIF":5.3000,"publicationDate":"2024-07-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11267579/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.4c00564","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/4 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
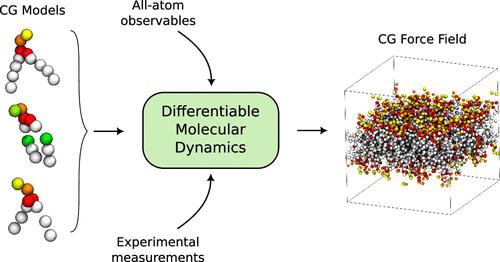
We develop ∂-HylleraasMD (∂-HyMD), a fully end-to-end differentiable molecular dynamics software based on the Hamiltonian hybrid particle-field formalism, and use it to establish a protocol for automated optimization of force field parameters. ∂-HyMD is templated on the recently released HylleraaasMD software, while using the JAX autodiff framework as the main engine for the differentiable dynamics. ∂-HyMD exploits an embarrassingly parallel optimization algorithm by spawning independent simulations, whose trajectories are simultaneously processed by reverse mode automatic differentiation to calculate the gradient of the loss function, which is in turn used for iterative optimization of the force-field parameters. We show that parallel organization facilitates the convergence of the minimization procedure, avoiding the known memory and numerical stability issues of differentiable molecular dynamics approaches. We showcase the effectiveness of our implementation by producing a library of force field parameters for standard phospholipids, with either zwitterionic or anionic heads and with saturated or unsaturated tails. Compared to the all-atom reference, the force field obtained by ∂-HyMD yields better density profiles than the parameters derived from previously utilized gradient-free optimization procedures. Moreover, ∂-HyMD models can predict with good accuracy properties not included in the learning objective, such as lateral pressure profiles, and are transferable to other systems, including triglycerides.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
