{"title":"A λ-Dynamics Investigation of Insulin <i>Wakayama</i> and Other A3 Variant Binding Affinities to the Insulin Receptor.","authors":"Monica P Barron, Jonah Z Vilseck","doi":"10.1021/acs.jcim.4c00662","DOIUrl":null,"url":null,"abstract":"<p><p>Insulin <i>Wakayama</i> is a clinical insulin variant where a conserved valine at the third residue on insulin's A chain (Val<sup>A3</sup>) is replaced with a leucine (Leu<sup>A3</sup>), weakening insulin receptor (IR) binding by 140-500-fold. This severe impact on binding from a subtle modification has posed an intriguing problem for decades. Although experimental investigations of natural and unnatural A3 mutations have highlighted the sensitivity of insulin-IR binding at this site, atomistic explanations of these binding trends have remained elusive. We investigate this problem computationally using λ-dynamics free energy calculations to model structural changes in response to perturbations of the Val<sup>A3</sup> side chain and to calculate associated relative changes in binding free energy (ΔΔ<i>G</i><sub>bind</sub>). The <i>Wakayama</i> Leu<sup>A3</sup> mutation and seven other A3 substitutions were studied in this work. The calculated ΔΔ<i>G</i><sub>bind</sub> results showed high agreement compared to experimental binding potencies with a Pearson correlation of 0.88 and a mean unsigned error of 0.68 kcal/mol. Extensive structural analyses of λ-dynamics trajectories revealed that critical interactions were disrupted between insulin and the insulin receptor as a result of the A3 mutations. This investigation also quantifies the effect that adding an A3 C<sub>δ</sub> atom or losing an A3 C<sub>γ</sub> atom has on insulin's binding affinity to the IR. Thus, λ-dynamics was able to successfully model the effects of mutations to insulin's A3 side chain on its protein-protein interactions with the IR and shed new light on a decades-old mystery: the exquisite sensitivity of hormone-receptor binding to a subtle modification of an invariant insulin residue.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":" ","pages":"5657-5670"},"PeriodicalIF":5.3000,"publicationDate":"2024-07-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11268370/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.4c00662","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/4 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
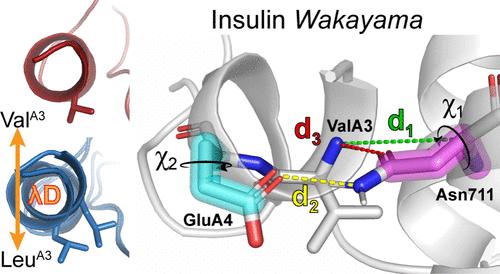
Insulin Wakayama is a clinical insulin variant where a conserved valine at the third residue on insulin's A chain (ValA3) is replaced with a leucine (LeuA3), weakening insulin receptor (IR) binding by 140-500-fold. This severe impact on binding from a subtle modification has posed an intriguing problem for decades. Although experimental investigations of natural and unnatural A3 mutations have highlighted the sensitivity of insulin-IR binding at this site, atomistic explanations of these binding trends have remained elusive. We investigate this problem computationally using λ-dynamics free energy calculations to model structural changes in response to perturbations of the ValA3 side chain and to calculate associated relative changes in binding free energy (ΔΔGbind). The Wakayama LeuA3 mutation and seven other A3 substitutions were studied in this work. The calculated ΔΔGbind results showed high agreement compared to experimental binding potencies with a Pearson correlation of 0.88 and a mean unsigned error of 0.68 kcal/mol. Extensive structural analyses of λ-dynamics trajectories revealed that critical interactions were disrupted between insulin and the insulin receptor as a result of the A3 mutations. This investigation also quantifies the effect that adding an A3 Cδ atom or losing an A3 Cγ atom has on insulin's binding affinity to the IR. Thus, λ-dynamics was able to successfully model the effects of mutations to insulin's A3 side chain on its protein-protein interactions with the IR and shed new light on a decades-old mystery: the exquisite sensitivity of hormone-receptor binding to a subtle modification of an invariant insulin residue.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
