{"title":"Structural characterization and keto-enol tautomerization of 4-substituted pyrazolone derivatives with DFT approach","authors":"Serpil Eryilmaz , Emine Bagdatli","doi":"10.1016/j.jmgm.2024.108814","DOIUrl":null,"url":null,"abstract":"<div><p>The synthesis of two pyrazolone derivative compounds, <strong>PYR-I</strong> <em>(4-Acetyl-1-(4-chlorophenyl)-3-isopropyl-1H-pyrazol-5(4H)-one)</em> and <strong>PYR-II</strong> <em>1-(4-Chlorophenyl))-3-isopropyl-5-oxo-4,5-5-dihydro-1H-pyrazole-4-carbaldehyde</em>, their characterization by FT-IR, NMR, UV–Vis and GC-MS techniques, and the evaluation of the keto-enol tautomerization process of the structures along with the DFT approach and spectral data were reported in this paper. Spectral findings indicated that <strong>PYR-I</strong> was stable at the keto state. The IR spectrum recorded in solid form showed that the <strong>PYR-II</strong> structure was stable in the enol state, while the NMR spectrum in the solution medium showed that it was stable in the keto state. DFT-based analyses were realized with the B3LYP hybrid functional and the 6–311++G(d,p) basis set. The modelled keto, transition and enol state molecular geometries of structures were optimized in the gas phase and different solvent media and the total energy and dipole moment values were investigated at the specified theoretical level. The possible keto-enol tautomerism mechanism of the structures was evaluated through some thermodynamic parameters such as the difference in free Gibbs energy (ΔG), enthalpy (ΔH), entropy (ΔS), and predictive tautomeric equilibrium constants (K<sub>eq</sub>), acidity constants (p<em>K</em><sub>a</sub>) and percentages of tautomers at 298.15 K and 1 atm pressure. The results of these analyses based on the DFT approach indicated that the keto-enol tautomer equilibrium heavily favours the keto form for <strong>PYR-I</strong> and the enol form for <strong>PYR-II</strong> in all cases. Moreover, natural bond orbital (NBO) analysis was performed for the tautomers, and the chemical reactivity profiles of the most stable tautomers were examined with the values of frontier molecular orbital energy and some reactivity descriptors.</p></div>","PeriodicalId":16361,"journal":{"name":"Journal of molecular graphics & modelling","volume":"131 ","pages":"Article 108814"},"PeriodicalIF":3.0000,"publicationDate":"2024-06-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular graphics & modelling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1093326324001141","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
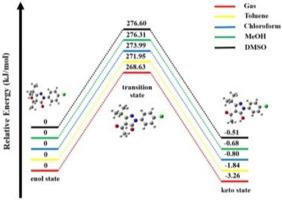
The synthesis of two pyrazolone derivative compounds, PYR-I(4-Acetyl-1-(4-chlorophenyl)-3-isopropyl-1H-pyrazol-5(4H)-one) and PYR-II1-(4-Chlorophenyl))-3-isopropyl-5-oxo-4,5-5-dihydro-1H-pyrazole-4-carbaldehyde, their characterization by FT-IR, NMR, UV–Vis and GC-MS techniques, and the evaluation of the keto-enol tautomerization process of the structures along with the DFT approach and spectral data were reported in this paper. Spectral findings indicated that PYR-I was stable at the keto state. The IR spectrum recorded in solid form showed that the PYR-II structure was stable in the enol state, while the NMR spectrum in the solution medium showed that it was stable in the keto state. DFT-based analyses were realized with the B3LYP hybrid functional and the 6–311++G(d,p) basis set. The modelled keto, transition and enol state molecular geometries of structures were optimized in the gas phase and different solvent media and the total energy and dipole moment values were investigated at the specified theoretical level. The possible keto-enol tautomerism mechanism of the structures was evaluated through some thermodynamic parameters such as the difference in free Gibbs energy (ΔG), enthalpy (ΔH), entropy (ΔS), and predictive tautomeric equilibrium constants (Keq), acidity constants (pKa) and percentages of tautomers at 298.15 K and 1 atm pressure. The results of these analyses based on the DFT approach indicated that the keto-enol tautomer equilibrium heavily favours the keto form for PYR-I and the enol form for PYR-II in all cases. Moreover, natural bond orbital (NBO) analysis was performed for the tautomers, and the chemical reactivity profiles of the most stable tautomers were examined with the values of frontier molecular orbital energy and some reactivity descriptors.
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
