Mokshi Sharma, Subrata Banik* and Tapta Kanchan Roy*,
{"title":"Performance of Effective Harmonic Oscillator Approach for the Calculations of Vibrational Transition Energies of Large Molecules","authors":"Mokshi Sharma, Subrata Banik* and Tapta Kanchan Roy*, ","doi":"10.1021/acs.jpca.4c01583","DOIUrl":null,"url":null,"abstract":"<p >The accuracy and performance of the effective harmonic oscillator approximation for the description of anharmonic vibrational structure calculations are tested for large molecular systems and compared with experimental values along with vibrational self-consistent field and second-order perturbation theories. The effective harmonic oscillator approach is an effective single-particle approximation where the variational parameters are the centroids and widths of the multidimensional Gaussian product functions posited as the vibrational wave functions. A comprehensive calculation for 849 transitions that include the fundamentals, two and three quanta overtone transitions, and several combination bands of three polyaromatic hydrocarbons and one DNA nucleobase with a total of 231 normal modes are assessed. A comparison of EHO results with the experimental values is done for the polyaromatic hydrocarbons, and a close agreement is found between the two results. It also offers anharmonic eigenstates and eigenfunctions that are nearly identical with vibrational self-consistent field theory. An extensive analysis on the resultant wave functions of the excited states is performed. The overall root-mean-square deviation (RMSD) between these two methods for 849 transitions understudy is only about 8.3 cm<sup>–1</sup>, suggesting the effective harmonic oscillator as a viable alternative for the reliable calculations of transition energies of large molecular systems.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":"128 28","pages":"5762–5776"},"PeriodicalIF":2.8000,"publicationDate":"2024-07-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpca.4c01583","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
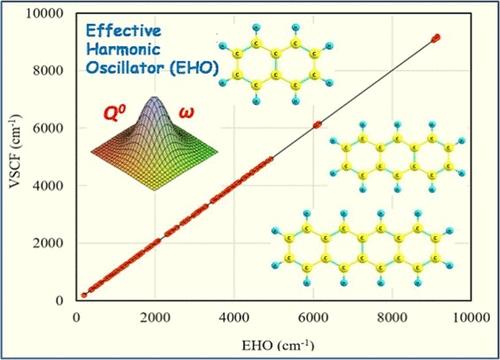
The accuracy and performance of the effective harmonic oscillator approximation for the description of anharmonic vibrational structure calculations are tested for large molecular systems and compared with experimental values along with vibrational self-consistent field and second-order perturbation theories. The effective harmonic oscillator approach is an effective single-particle approximation where the variational parameters are the centroids and widths of the multidimensional Gaussian product functions posited as the vibrational wave functions. A comprehensive calculation for 849 transitions that include the fundamentals, two and three quanta overtone transitions, and several combination bands of three polyaromatic hydrocarbons and one DNA nucleobase with a total of 231 normal modes are assessed. A comparison of EHO results with the experimental values is done for the polyaromatic hydrocarbons, and a close agreement is found between the two results. It also offers anharmonic eigenstates and eigenfunctions that are nearly identical with vibrational self-consistent field theory. An extensive analysis on the resultant wave functions of the excited states is performed. The overall root-mean-square deviation (RMSD) between these two methods for 849 transitions understudy is only about 8.3 cm–1, suggesting the effective harmonic oscillator as a viable alternative for the reliable calculations of transition energies of large molecular systems.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
