{"title":"Ab initio exploration of low-lying electronic states of linear and bent MNX+ (M = Ca, Sr, Ba, Ra; X = O, S, Se, Te, Po) and their origins","authors":"Isuru R. Ariyarathna","doi":"10.1002/jcc.27456","DOIUrl":null,"url":null,"abstract":"<p>High-level multireference and coupled cluster quantum calculations were employed to analyze low-lying electronic states of linear-MNX<sup>+</sup> and side-bonded-M[NX]<sup>+</sup> (M = Ca, Sr, Ba, Ra; X = O, S, Se, Te, Po) species. Their full potential energy curves (PECs), dissociation energies (<i>D<sub>e</sub></i>s), geometric parameters, excitation energies (<i>T</i><sub><i>e</i></sub>s), and harmonic vibrational frequencies (<i>ω</i><sub><i>e</i></sub>s) are reported. The first three chemically bound electronic states of MNX<sup>+</sup> and M[NX]<sup>+</sup> are <sup>3</sup>∑<sup>−</sup>, <sup>1</sup>Δ, <sup>1</sup>∑<sup>+</sup> and <sup>3</sup>A″, <sup>1</sup>A′, <sup>1</sup>A″, respectively. The <sup>3</sup>∑<sup>−</sup>, <sup>1</sup>Δ, <sup>1</sup>∑<sup>+</sup> of MNX<sup>+</sup> originate from the M<sup>+</sup>(<sup>2</sup>D) + NX(<sup>2</sup>Π) fragments, whereas the <sup>3</sup>A″, <sup>1</sup>A′, <sup>1</sup>A″ states of M[NX]<sup>+</sup> dissociate to M<sup>+</sup>(<sup>2</sup>S) + NX(<sup>2</sup>Π) as a result of avoided crossings. The MNX<sup>+</sup> and M[NX]<sup>+</sup> are real minima on the potential energy surface and their interconversions are possible. The M<sup>2+</sup>NX<sup>−</sup>/M<sup>2+</sup>[NX]<sup>−</sup> ionic structure is an accurate representation for their low-lying electronic states. The <i>D</i><sub>e</sub>s of MNX<sup>+</sup> species were found to depend on the dipole moment (<i>μ</i>) of the corresponding NX ligands and a linear relationship between these two parameters was observed.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 30","pages":"2530-2538"},"PeriodicalIF":4.8000,"publicationDate":"2024-07-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27456","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27456","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
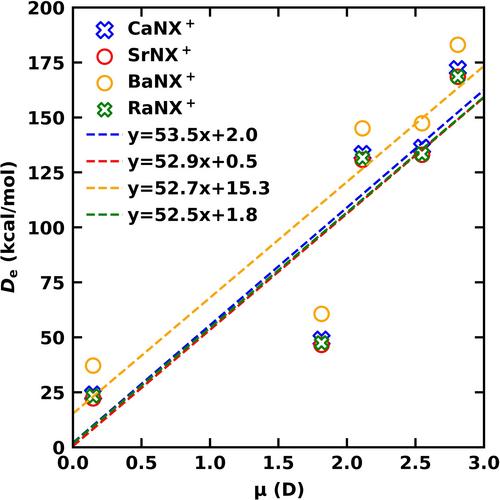
High-level multireference and coupled cluster quantum calculations were employed to analyze low-lying electronic states of linear-MNX+ and side-bonded-M[NX]+ (M = Ca, Sr, Ba, Ra; X = O, S, Se, Te, Po) species. Their full potential energy curves (PECs), dissociation energies (Des), geometric parameters, excitation energies (Tes), and harmonic vibrational frequencies (ωes) are reported. The first three chemically bound electronic states of MNX+ and M[NX]+ are 3∑−, 1Δ, 1∑+ and 3A″, 1A′, 1A″, respectively. The 3∑−, 1Δ, 1∑+ of MNX+ originate from the M+(2D) + NX(2Π) fragments, whereas the 3A″, 1A′, 1A″ states of M[NX]+ dissociate to M+(2S) + NX(2Π) as a result of avoided crossings. The MNX+ and M[NX]+ are real minima on the potential energy surface and their interconversions are possible. The M2+NX−/M2+[NX]− ionic structure is an accurate representation for their low-lying electronic states. The Des of MNX+ species were found to depend on the dipole moment (μ) of the corresponding NX ligands and a linear relationship between these two parameters was observed.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
