Development and implementation of a core genome multilocus sequence typing scheme for Yersinia enterocolitica: a tool for surveillance and outbreak detection.
{"title":"Development and implementation of a core genome multilocus sequence typing scheme for <i>Yersinia enterocolitica:</i> a tool for surveillance and outbreak detection.","authors":"Joao Pires, Lin T Brandal, Umaer Naseer","doi":"10.1128/jcm.00040-24","DOIUrl":null,"url":null,"abstract":"<p><p><i>Yersinia enterocolitica</i> (<i>Y. enterocolitica</i>) is the most frequent etiological agent of yersiniosis and has been responsible for several national outbreaks in Norway and elsewhere. A standardized high-resolution method, such as core genome Multilocus Sequence Typing (cgMLST), is needed for pathogen traceability at the national and international levels. In this study, we developed and implemented a cgMLST scheme for <i>Y. enterocolitica</i>. We designed a cgMLST scheme in SeqSphere + using high-quality genomes from different <i>Y. enterocolitica</i> biotype sublineages. The scheme was validated if more than 95% of targets were found across all tested <i>Y. enterocolitica</i>: 563 Norwegian genomes collected between 2012 and 2022 and 327 genomes from public data sets. We applied the scheme to known outbreaks to establish a threshold for identifying major complex types (CTs) based on the number of allelic differences. The final cgMLST scheme included 2,582 genes with a median of 97.9% (interquartile range 97.6%-98.8%) targets found across all tested genomes. Analysis of outbreaks identified all outbreak strains using single linkage clustering at four allelic differences. This threshold identified 311 unique CTs in Norway, of which CT18, CT12, and CT5 were identified as the most frequently associated with outbreaks. The cgMLST scheme showed a very good performance in typing <i>Y. enterocolitica</i> using diverse data sources and was able to identify outbreak clusters. We recommend the implementation of this scheme nationally and internationally to facilitate <i>Y. enterocolitica</i> surveillance and improve outbreak response in national and cross-border outbreaks.</p>","PeriodicalId":15511,"journal":{"name":"Journal of Clinical Microbiology","volume":" ","pages":"e0004024"},"PeriodicalIF":5.4000,"publicationDate":"2024-08-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11325262/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Clinical Microbiology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1128/jcm.00040-24","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/11 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"MICROBIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
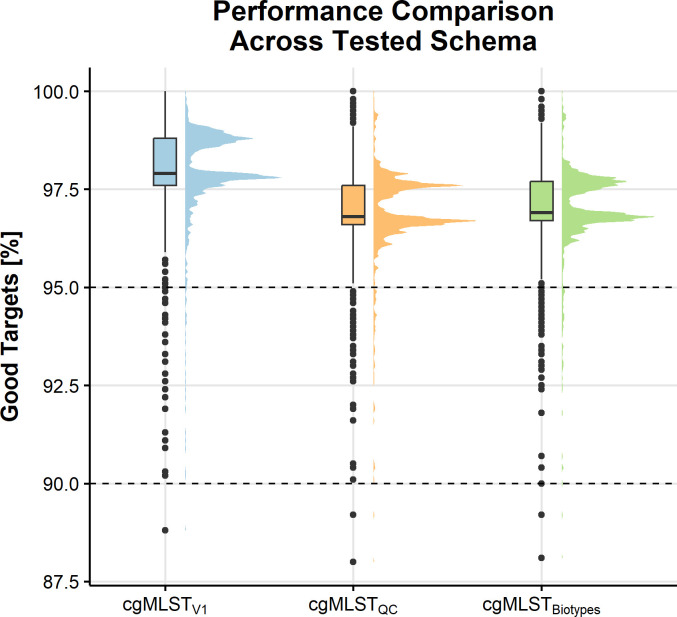
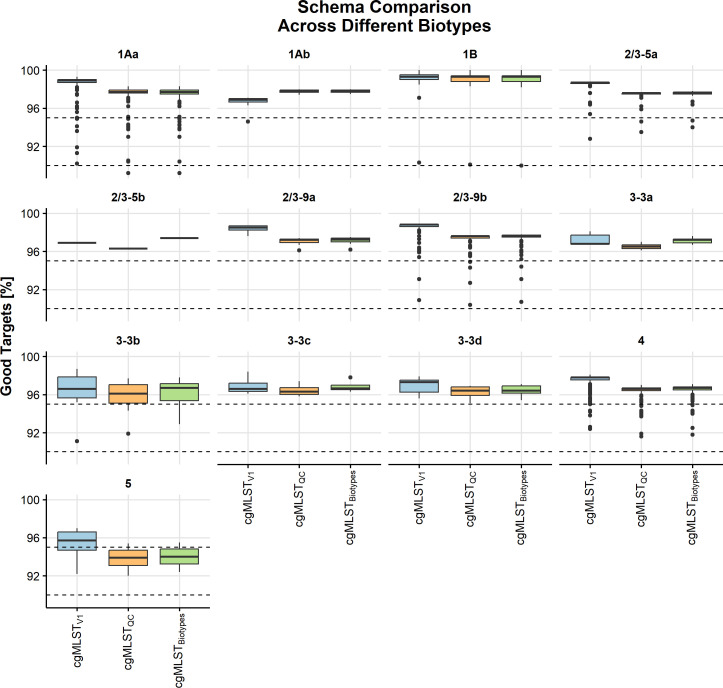
Yersinia enterocolitica (Y. enterocolitica) is the most frequent etiological agent of yersiniosis and has been responsible for several national outbreaks in Norway and elsewhere. A standardized high-resolution method, such as core genome Multilocus Sequence Typing (cgMLST), is needed for pathogen traceability at the national and international levels. In this study, we developed and implemented a cgMLST scheme for Y. enterocolitica. We designed a cgMLST scheme in SeqSphere + using high-quality genomes from different Y. enterocolitica biotype sublineages. The scheme was validated if more than 95% of targets were found across all tested Y. enterocolitica: 563 Norwegian genomes collected between 2012 and 2022 and 327 genomes from public data sets. We applied the scheme to known outbreaks to establish a threshold for identifying major complex types (CTs) based on the number of allelic differences. The final cgMLST scheme included 2,582 genes with a median of 97.9% (interquartile range 97.6%-98.8%) targets found across all tested genomes. Analysis of outbreaks identified all outbreak strains using single linkage clustering at four allelic differences. This threshold identified 311 unique CTs in Norway, of which CT18, CT12, and CT5 were identified as the most frequently associated with outbreaks. The cgMLST scheme showed a very good performance in typing Y. enterocolitica using diverse data sources and was able to identify outbreak clusters. We recommend the implementation of this scheme nationally and internationally to facilitate Y. enterocolitica surveillance and improve outbreak response in national and cross-border outbreaks.
期刊介绍:
The Journal of Clinical Microbiology® disseminates the latest research concerning the laboratory diagnosis of human and animal infections, along with the laboratory's role in epidemiology and the management of infectious diseases.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
