{"title":"Citrin deficiency—The East-side story","authors":"Johannes Häberle","doi":"10.1002/jimd.12772","DOIUrl":null,"url":null,"abstract":"<p>Citrin deficiency (CD) is a complex metabolic condition due to defects in <i>SLC25A13</i> encoding citrin, an aspartate/glutamate carrier located in the mitochondrial inner membrane. The condition was first described in Japan and other East Asian countries in patients who were thought to suffer from classical citrullinemia type 1, and was therefore classified as a urea cycle disorder. With an improved understanding of its molecular basis, it became apparent that a defect of citrin is primarily affecting the malate–aspartate shuttle with however multiple secondary effects on many central metabolic pathways including glycolysis, gluconeogenesis, de novo lipogenesis and ureagenesis. In the meantime, it became also clear that CD must be considered as a global disease with patients identified in many parts of the world and affected by <i>SLC25A13</i> genotypes different from those known in East Asian populations. The present short review summarizes the (hi)story of this complex metabolic condition and tries to explain the relevance of including CD as a differential diagnosis in neonates and infants with cholestasis and in (not only adult) patients with hyperammonemia of unknown origin with subsequent impact on the emergency management.</p>","PeriodicalId":16281,"journal":{"name":"Journal of Inherited Metabolic Disease","volume":"47 6","pages":"1129-1133"},"PeriodicalIF":4.2000,"publicationDate":"2024-07-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jimd.12772","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Inherited Metabolic Disease","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jimd.12772","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
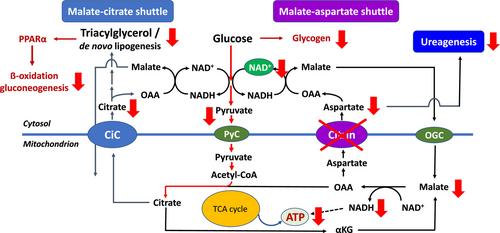
Citrin deficiency (CD) is a complex metabolic condition due to defects in SLC25A13 encoding citrin, an aspartate/glutamate carrier located in the mitochondrial inner membrane. The condition was first described in Japan and other East Asian countries in patients who were thought to suffer from classical citrullinemia type 1, and was therefore classified as a urea cycle disorder. With an improved understanding of its molecular basis, it became apparent that a defect of citrin is primarily affecting the malate–aspartate shuttle with however multiple secondary effects on many central metabolic pathways including glycolysis, gluconeogenesis, de novo lipogenesis and ureagenesis. In the meantime, it became also clear that CD must be considered as a global disease with patients identified in many parts of the world and affected by SLC25A13 genotypes different from those known in East Asian populations. The present short review summarizes the (hi)story of this complex metabolic condition and tries to explain the relevance of including CD as a differential diagnosis in neonates and infants with cholestasis and in (not only adult) patients with hyperammonemia of unknown origin with subsequent impact on the emergency management.
期刊介绍:
The Journal of Inherited Metabolic Disease (JIMD) is the official journal of the Society for the Study of Inborn Errors of Metabolism (SSIEM). By enhancing communication between workers in the field throughout the world, the JIMD aims to improve the management and understanding of inherited metabolic disorders. It publishes results of original research and new or important observations pertaining to any aspect of inherited metabolic disease in humans and higher animals. This includes clinical (medical, dental and veterinary), biochemical, genetic (including cytogenetic, molecular and population genetic), experimental (including cell biological), methodological, theoretical, epidemiological, ethical and counselling aspects. The JIMD also reviews important new developments or controversial issues relating to metabolic disorders and publishes reviews and short reports arising from the Society''s annual symposia. A distinction is made between peer-reviewed scientific material that is selected because of its significance for other professionals in the field and non-peer- reviewed material that aims to be important, controversial, interesting or entertaining (“Extras”).



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
