{"title":"PARP1-driven repair of topoisomerase IIIα DNA-protein crosslinks by FEN1.","authors":"Liton Kumar Saha, Yilun Sun, Sourav Saha, Xi Yang, Yves Pommier","doi":"10.1016/j.celrep.2024.114522","DOIUrl":null,"url":null,"abstract":"<p><p>Persistent DNA-protein crosslinks formed by human topoisomerase IIIα (TOP3A-DPCs) interfere with DNA metabolism and lead to genome damage and cell death. Recently, we demonstrated that such abortive TOP3A-DPCs are ubiquitylated and proteolyzed by Spartan (SPRTN). Here, we identify transient poly(ADP-ribosylation) (PARylation) in addition to ubiquitylation as a signaling mechanism for TOP3A-DPC repair and provide evidence that poly(ADP-ribose) polymerase 1 (PARP1) drives the repair of TOP3A-DPCs by recruiting flap endonuclease 1 (FEN1) to the TOP3A-DPCs. We find that blocking PARylation attenuates the interaction of FEN1 and TOP3A and that TOP3A-DPCs accumulate in cells with compromised PARP1 activity and in FEN1-deficient cells. We also show that PARP1 suppresses TOP3A-DPC ubiquitylation and that inhibiting the ubiquitin-activating enzyme E1 (UBE1) increases TOP3A-DPCs, consistent with ubiquitylation serving as a signaling mechanism for TOP3A-DPC repair mediated by SPRTN and TDP2. We propose that two concerted pathways repair TOP3A-DPCs: PARylation-driven FEN1 excision and ubiquitylation-driven SPRTN-TDP2 excision.</p>","PeriodicalId":9798,"journal":{"name":"Cell reports","volume":"43 8","pages":"114522"},"PeriodicalIF":6.9000,"publicationDate":"2024-08-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11513513/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cell reports","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1016/j.celrep.2024.114522","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/18 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 0
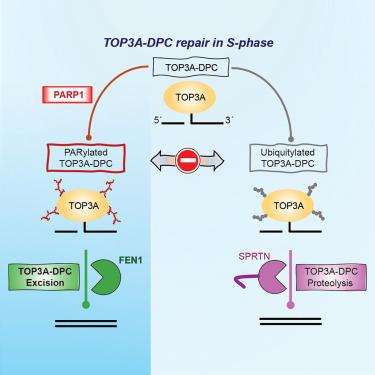
Abstract
Persistent DNA-protein crosslinks formed by human topoisomerase IIIα (TOP3A-DPCs) interfere with DNA metabolism and lead to genome damage and cell death. Recently, we demonstrated that such abortive TOP3A-DPCs are ubiquitylated and proteolyzed by Spartan (SPRTN). Here, we identify transient poly(ADP-ribosylation) (PARylation) in addition to ubiquitylation as a signaling mechanism for TOP3A-DPC repair and provide evidence that poly(ADP-ribose) polymerase 1 (PARP1) drives the repair of TOP3A-DPCs by recruiting flap endonuclease 1 (FEN1) to the TOP3A-DPCs. We find that blocking PARylation attenuates the interaction of FEN1 and TOP3A and that TOP3A-DPCs accumulate in cells with compromised PARP1 activity and in FEN1-deficient cells. We also show that PARP1 suppresses TOP3A-DPC ubiquitylation and that inhibiting the ubiquitin-activating enzyme E1 (UBE1) increases TOP3A-DPCs, consistent with ubiquitylation serving as a signaling mechanism for TOP3A-DPC repair mediated by SPRTN and TDP2. We propose that two concerted pathways repair TOP3A-DPCs: PARylation-driven FEN1 excision and ubiquitylation-driven SPRTN-TDP2 excision.
期刊介绍:
Cell Reports publishes high-quality research across the life sciences and focuses on new biological insight as its primary criterion for publication. The journal offers three primary article types: Reports, which are shorter single-point articles, research articles, which are longer and provide deeper mechanistic insights, and resources, which highlight significant technical advances or major informational datasets that contribute to biological advances. Reviews covering recent literature in emerging and active fields are also accepted.
The Cell Reports Portfolio includes gold open-access journals that cover life, medical, and physical sciences, and its mission is to make cutting-edge research and methodologies available to a wide readership.
The journal's professional in-house editors work closely with authors, reviewers, and the scientific advisory board, which consists of current and future leaders in their respective fields. The advisory board guides the scope, content, and quality of the journal, but editorial decisions are independently made by the in-house scientific editors of Cell Reports.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
