Yajing Wang , Jianan Yao , Qingxiao Zhou , Dongtao Su , Weiwei Ju , Leyan Wang
{"title":"Transition-metal doped Ti2CO2 as gas sensor toward NH3: A DFT study","authors":"Yajing Wang , Jianan Yao , Qingxiao Zhou , Dongtao Su , Weiwei Ju , Leyan Wang","doi":"10.1016/j.chemphys.2024.112376","DOIUrl":null,"url":null,"abstract":"<div><p>MXenes is a novel material that has potential to be widely used in indoor hazardous gas detection. Based on the density functional theory (DFT), adsorption properties of NH<sub>3</sub> molecules on perfect, O-vacancy, transition-metal doped (TMs = Sc, V, Cr, Mn, Co, Ni) Ti<sub>2</sub>CO<sub>2</sub> substrates were investigated. The results showed that the adsorption of NH<sub>3</sub> molecules on the perfect and defective Ti<sub>2</sub>CO<sub>2</sub> was physical adsorption. After introducing of O-vacancy and dopant, the adsorption of NH<sub>3</sub> on the TM-doped substrates changed to be chemisorption. Interestingly, the V- and Ni-doped Ti<sub>2</sub>CO<sub>2</sub> exhibited obvious band gap change before and after the adsorption of NH<sub>3</sub> molecule, which could be used as an electrical signal to detect NH<sub>3</sub> gas. In addition, the results of density of states suggested that the enhancement of adsorption stability for NH<sub>3</sub> molecule was due to the hybridization between the 3d orbital of dopants and s (p) orbital of NH<sub>3</sub>. These results are expected to provide ideas for the design of NH<sub>3</sub> gas sensors based on MXenes materials.</p></div>","PeriodicalId":272,"journal":{"name":"Chemical Physics","volume":"586 ","pages":"Article 112376"},"PeriodicalIF":2.4000,"publicationDate":"2024-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0301010424002052","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/18 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
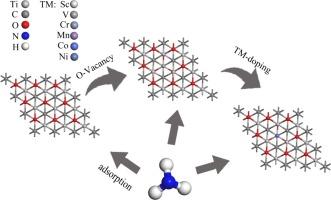
MXenes is a novel material that has potential to be widely used in indoor hazardous gas detection. Based on the density functional theory (DFT), adsorption properties of NH3 molecules on perfect, O-vacancy, transition-metal doped (TMs = Sc, V, Cr, Mn, Co, Ni) Ti2CO2 substrates were investigated. The results showed that the adsorption of NH3 molecules on the perfect and defective Ti2CO2 was physical adsorption. After introducing of O-vacancy and dopant, the adsorption of NH3 on the TM-doped substrates changed to be chemisorption. Interestingly, the V- and Ni-doped Ti2CO2 exhibited obvious band gap change before and after the adsorption of NH3 molecule, which could be used as an electrical signal to detect NH3 gas. In addition, the results of density of states suggested that the enhancement of adsorption stability for NH3 molecule was due to the hybridization between the 3d orbital of dopants and s (p) orbital of NH3. These results are expected to provide ideas for the design of NH3 gas sensors based on MXenes materials.
期刊介绍:
Chemical Physics publishes experimental and theoretical papers on all aspects of chemical physics. In this journal, experiments are related to theory, and in turn theoretical papers are related to present or future experiments. Subjects covered include: spectroscopy and molecular structure, interacting systems, relaxation phenomena, biological systems, materials, fundamental problems in molecular reactivity, molecular quantum theory and statistical mechanics. Computational chemistry studies of routine character are not appropriate for this journal.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
