Xuan Zhang, Qi-Jun Liu, Fu-Sheng Liu, Zheng-Tang Liu
{"title":"Predicting the thermal decomposition temperature of energetic materials from a simple model","authors":"Xuan Zhang, Qi-Jun Liu, Fu-Sheng Liu, Zheng-Tang Liu","doi":"10.1007/s00894-024-06075-z","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>The key factor in designing heat-resistant energetic materials is their thermal sensitivity. Further research and prediction of thermal sensitivity remains a great challenge for us. This study is based on first-principles calculations and establishes a theoretical model, which comprehensively considers band gap, density of states, and Young's modulus to obtain a empirical parameter Ψ. A quantitative relationship was established between the new parameter and the thermal decomposition temperature. The value of Ψ is calculated for 10 energetic materials and is found to have a strong correlation with the experimental thermal decomposition temperature. This further proves the reliability of our model. Specifically, the larger the value of Ψ, the higher the thermal decomposition temperature, and the more stable the energetic material will be. Therefore, to some extent, we can use the new parameter Ψ calculated by the model to predict thermal sensitivity.</p><h3>Methods</h3><p>Based on first-principles, this paper used the Cambridge Serial Total Energy Package (CASTEP) module of Materials Studio (MS) for calculations. The Perdew-Burke-Ernzerhof (PBE) functionals in Generalized Gradient Approximation (GGA) method as well as the Grimme dispersion correction was used in this paper.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"30 8","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2024-07-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-024-06075-z","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Context
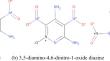
The key factor in designing heat-resistant energetic materials is their thermal sensitivity. Further research and prediction of thermal sensitivity remains a great challenge for us. This study is based on first-principles calculations and establishes a theoretical model, which comprehensively considers band gap, density of states, and Young's modulus to obtain a empirical parameter Ψ. A quantitative relationship was established between the new parameter and the thermal decomposition temperature. The value of Ψ is calculated for 10 energetic materials and is found to have a strong correlation with the experimental thermal decomposition temperature. This further proves the reliability of our model. Specifically, the larger the value of Ψ, the higher the thermal decomposition temperature, and the more stable the energetic material will be. Therefore, to some extent, we can use the new parameter Ψ calculated by the model to predict thermal sensitivity.
Methods
Based on first-principles, this paper used the Cambridge Serial Total Energy Package (CASTEP) module of Materials Studio (MS) for calculations. The Perdew-Burke-Ernzerhof (PBE) functionals in Generalized Gradient Approximation (GGA) method as well as the Grimme dispersion correction was used in this paper.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
