{"title":"Hakai, a novel Runx2 interacting protein, augments osteoblast differentiation by rescuing Runx2 from Smurf2-mediated proteasome degradation","authors":"Vishal Upadhyay, Shivani Sharma, Arppita Sethi, Anil Kumar Singh, Sangita Chowdhury, Swati Srivastava, Shivkant Mishra, Shyam Singh, Naibedya Chattopadhyay, Arun Kumar Trivedi","doi":"10.1002/jcp.31388","DOIUrl":null,"url":null,"abstract":"<p>Runt-related transcription factor 2 (Runx2) is a key regulator of osteoblast differentiation and bone formation. In Runx2-deficient embryos, skeletal development ceases at the cartilage anlage stage. These embryos die of respiratory failure upon birth and display a complete absence of bone and cartilage mineralization. Here, we identified Hakai, a type of E3 ubiquitin ligase as a potential Runx2 interacting partner through affinity pulldown-based proteomic approach. Subsequently, we observed that similar to Runx2, Hakai was downregulated in osteopenic ovariectomized rats, suggesting its involvement in bone formation. Consistent with this observation, Hakai overexpression significantly enhanced osteoblast differentiation in mesenchyme-like C3H10T1/2 as well as primary rat calvaria osteoblast (RCO) cells in vitro. Conversely, overexpression of a catalytically inactive Hakai mutant (C109A) exhibited minimal to no effect, whereas Hakai depletion markedly reduced endogenous Runx2 levels and impaired osteogenic differentiation in both C3H10T1/2 and RCOs. Mechanistically, Hakai physically interacts with Runx2 and enhances its protein turnover by rescuing it from Smad ubiquitination regulatory factor 2 (Smurf2)-mediated proteasome degradation. Wild-type Hakai but not Hakai-C109A inhibited Smurf2 protein levels through proteasome-mediated degradation. These findings underscore Hakai's functional role in bone formation, primarily through its positive modulation of Runx2 protein turnover by protecting it from Smurf2-mediated ubiquitin-proteasomal degradation. Collectively, our results demonstrate Hakai as a promising novel therapeutic target for osteoporosis.</p>","PeriodicalId":15220,"journal":{"name":"Journal of Cellular Physiology","volume":"239 9","pages":""},"PeriodicalIF":4.0000,"publicationDate":"2024-07-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Cellular Physiology","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcp.31388","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 0
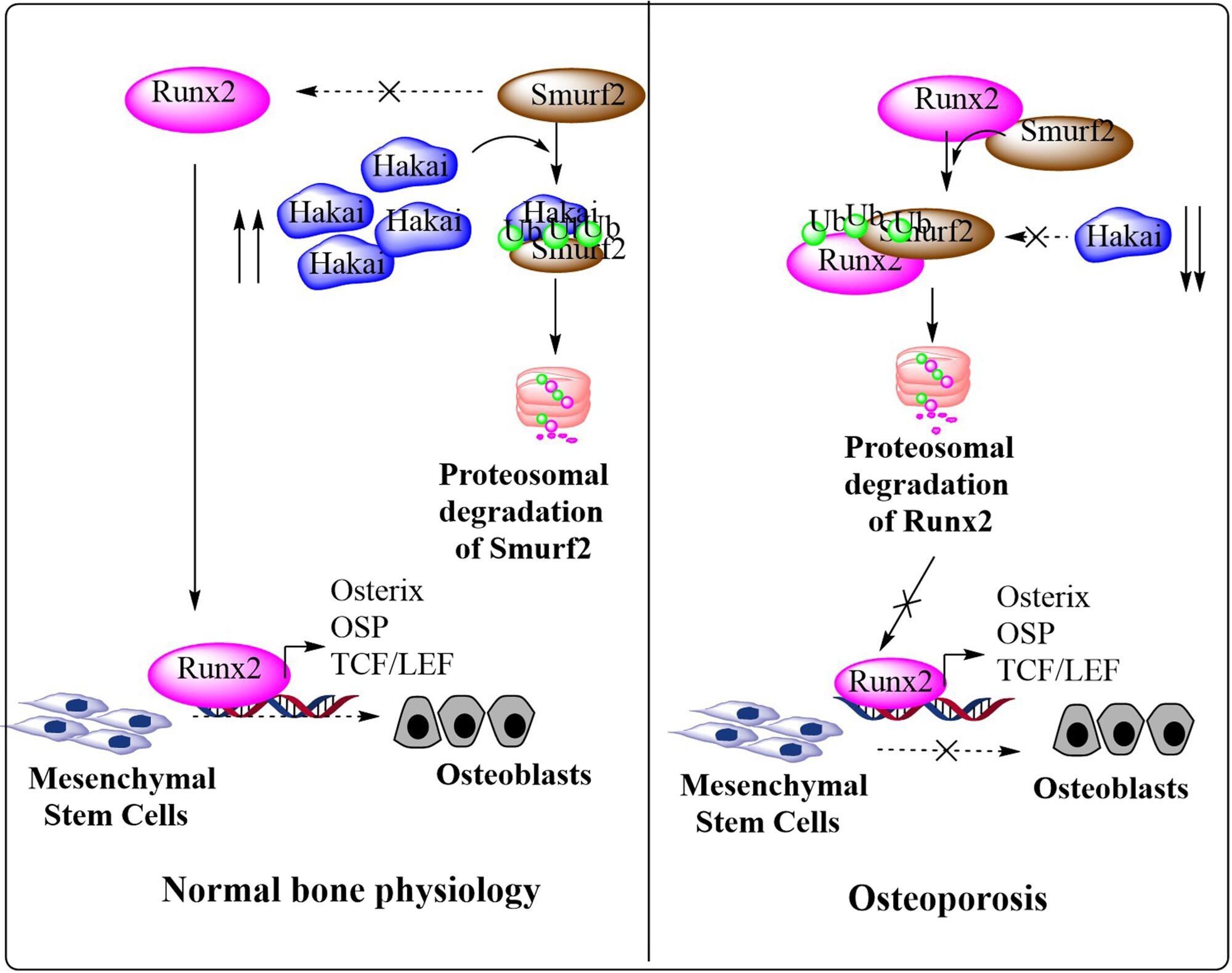
Abstract
Runt-related transcription factor 2 (Runx2) is a key regulator of osteoblast differentiation and bone formation. In Runx2-deficient embryos, skeletal development ceases at the cartilage anlage stage. These embryos die of respiratory failure upon birth and display a complete absence of bone and cartilage mineralization. Here, we identified Hakai, a type of E3 ubiquitin ligase as a potential Runx2 interacting partner through affinity pulldown-based proteomic approach. Subsequently, we observed that similar to Runx2, Hakai was downregulated in osteopenic ovariectomized rats, suggesting its involvement in bone formation. Consistent with this observation, Hakai overexpression significantly enhanced osteoblast differentiation in mesenchyme-like C3H10T1/2 as well as primary rat calvaria osteoblast (RCO) cells in vitro. Conversely, overexpression of a catalytically inactive Hakai mutant (C109A) exhibited minimal to no effect, whereas Hakai depletion markedly reduced endogenous Runx2 levels and impaired osteogenic differentiation in both C3H10T1/2 and RCOs. Mechanistically, Hakai physically interacts with Runx2 and enhances its protein turnover by rescuing it from Smad ubiquitination regulatory factor 2 (Smurf2)-mediated proteasome degradation. Wild-type Hakai but not Hakai-C109A inhibited Smurf2 protein levels through proteasome-mediated degradation. These findings underscore Hakai's functional role in bone formation, primarily through its positive modulation of Runx2 protein turnover by protecting it from Smurf2-mediated ubiquitin-proteasomal degradation. Collectively, our results demonstrate Hakai as a promising novel therapeutic target for osteoporosis.
期刊介绍:
The Journal of Cellular Physiology publishes reports of high biological significance in areas of eukaryotic cell biology and physiology, focusing on those articles that adopt a molecular mechanistic approach to investigate cell structure and function. There is appreciation for the application of cellular, biochemical, molecular and in vivo genetic approaches, as well as the power of genomics, proteomics, bioinformatics and systems biology. In particular, the Journal encourages submission of high-interest papers investigating the genetic and epigenetic regulation of proliferation and phenotype as well as cell fate and lineage commitment by growth factors, cytokines and their cognate receptors and signal transduction pathways that influence the expression, integration and activities of these physiological mediators. Similarly, the Journal encourages submission of manuscripts exploring the regulation of growth and differentiation by cell adhesion molecules in addition to the interplay between these processes and those induced by growth factors and cytokines. Studies on the genes and processes that regulate cell cycle progression and phase transition in eukaryotic cells, and the mechanisms that determine whether cells enter quiescence, proliferate or undergo apoptosis are also welcomed. Submission of papers that address contributions of the extracellular matrix to cellular phenotypes and physiological control as well as regulatory mechanisms governing fertilization, embryogenesis, gametogenesis, cell fate, lineage commitment, differentiation, development and dynamic parameters of cell motility are encouraged. Finally, the investigation of stem cells and changes that differentiate cancer cells from normal cells including studies on the properties and functions of oncogenes and tumor suppressor genes will remain as one of the major interests of the Journal.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
