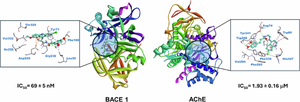
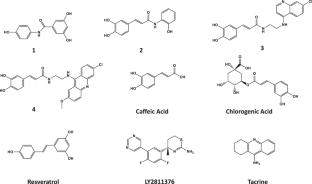
{"title":"Synthesis, in vitro activity, and molecular docking of caffeic acid and resveratrol derivatives against Alzheimer’s disease-related enzymes","authors":"Alberto Martínez","doi":"10.1007/s00044-024-03278-0","DOIUrl":null,"url":null,"abstract":"<div><p>Alzheimer’s disease (AD) is the most common form of dementia affecting about 40 million people around the world. The number of people living with this ailment is expected to triple in the next 50 years due to the aging population and the lack of any effective treatment. In this work we have synthesized a group of three hybrid caffeic acid and a resveratrol derivatives (<b>1-4</b>), and we have tested their ability to inhibit in vitro the enzymatic activity of the beta-site amyloid precursor protein cleaving protein enzyme 1 (BACE 1) and acetylcholinesterase (AChE). The inhibitory activity was compared to that of parent compounds caffeic acid and resveratrol, as well as related chlorogenic acid. Clinically tested LY2811376 and tacrine were used as positive controls. All three caffeic acid derivatives displayed better inhibitory activity than parent caffeic acid and chlorogenic acid. In particular, the in vitro IC<sub>50</sub> for compound <b>4</b> against BACE 1 fell in the nanomolar range (69 ± 5 nM), comparable or better than LY2811376 (173 ± 8 nM) which reached Phase I clinical trials against AD as a BACE 1 inhibitor. On the other hand, compound <b>3</b> showed a remarkable AChE inhibitory potency in the low micromolar range (1.93 ± 0.16 μM). Molecular docking was performed to gain valuable insights into the interactions between compounds <b>1-4</b> and the active sites of both BACE 1 and AChE. Calculated binding affinities generally correlated well with experimental in vitro inhibition. Experimental and molecular docking results validated the proposed drug design, since the most active compounds <b>3</b> and <b>4</b> established interactions with relevant amino acid residues of the BACE 1 and AChE active sites through the different pharmacophore features of the hybrid structures. Overall, the results presented in this work could potentially have important implications in the rational design of compounds with potential anti-AD properties.</p><div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>","PeriodicalId":699,"journal":{"name":"Medicinal Chemistry Research","volume":"33 9","pages":"1681 - 1697"},"PeriodicalIF":3.5000,"publicationDate":"2024-07-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Medicinal Chemistry Research","FirstCategoryId":"3","ListUrlMain":"https://link.springer.com/article/10.1007/s00044-024-03278-0","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
Alzheimer’s disease (AD) is the most common form of dementia affecting about 40 million people around the world. The number of people living with this ailment is expected to triple in the next 50 years due to the aging population and the lack of any effective treatment. In this work we have synthesized a group of three hybrid caffeic acid and a resveratrol derivatives (1-4), and we have tested their ability to inhibit in vitro the enzymatic activity of the beta-site amyloid precursor protein cleaving protein enzyme 1 (BACE 1) and acetylcholinesterase (AChE). The inhibitory activity was compared to that of parent compounds caffeic acid and resveratrol, as well as related chlorogenic acid. Clinically tested LY2811376 and tacrine were used as positive controls. All three caffeic acid derivatives displayed better inhibitory activity than parent caffeic acid and chlorogenic acid. In particular, the in vitro IC50 for compound 4 against BACE 1 fell in the nanomolar range (69 ± 5 nM), comparable or better than LY2811376 (173 ± 8 nM) which reached Phase I clinical trials against AD as a BACE 1 inhibitor. On the other hand, compound 3 showed a remarkable AChE inhibitory potency in the low micromolar range (1.93 ± 0.16 μM). Molecular docking was performed to gain valuable insights into the interactions between compounds 1-4 and the active sites of both BACE 1 and AChE. Calculated binding affinities generally correlated well with experimental in vitro inhibition. Experimental and molecular docking results validated the proposed drug design, since the most active compounds 3 and 4 established interactions with relevant amino acid residues of the BACE 1 and AChE active sites through the different pharmacophore features of the hybrid structures. Overall, the results presented in this work could potentially have important implications in the rational design of compounds with potential anti-AD properties.
期刊介绍:
Medicinal Chemistry Research (MCRE) publishes papers on a wide range of topics, favoring research with significant, new, and up-to-date information. Although the journal has a demanding peer review process, MCRE still boasts rapid publication, due in part, to the length of the submissions. The journal publishes significant research on various topics, many of which emphasize the structure-activity relationships of molecular biology.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
