Tail-approach based design, synthesis, and molecular modeling of benzenesulfonamides carrying thiadiazole and urea moieties as novel carbonic anhydrase inhibitors
M. İhsan Han, Miyase Gözde Gündüz, Andrea Ammara, Claudiu T. Supuran, Şengül Dilem Doğan
{"title":"Tail-approach based design, synthesis, and molecular modeling of benzenesulfonamides carrying thiadiazole and urea moieties as novel carbonic anhydrase inhibitors","authors":"M. İhsan Han, Miyase Gözde Gündüz, Andrea Ammara, Claudiu T. Supuran, Şengül Dilem Doğan","doi":"10.1002/ardp.202400439","DOIUrl":null,"url":null,"abstract":"<p>We synthesized herein 16 compounds (<b>SUT1</b>–<b>SUT16</b>) as potential carbonic anhydrase (CA) inhibitors utilizing the tail-approach design. Based on this strategy, we connected benzenesulfonamide, the zinc-binding scaffold, to different urea moieties with the 1,3,4-thiadiazole ring as a linker. We obtained the target compounds by the reaction of 4-(5-amino-1,3,4-thiadiazol-2-yl)benzenesulfonamide with aryl isocyanates. Upon confirmation of their structures, the compounds were screened for their ability to inhibit the tumor-related human (h) isoforms human carbonic anhydrase (hCA) IX and XII, as well as the physiologically dominant hCA I and II. Most of the molecules demonstrated <i>K</i><sub>i</sub> values ≤ 10 nM with different selectivity profiles. The binding modes of <b>SUT9</b>, <b>SUT10</b>, and <b>SUT5</b>, the most effective inhibitors of hCA II, IX, and XII, respectively, were predicted by molecular docking. <b>SUT16</b> (4-{5-[3-(naphthalen-1-yl)ureido]-1,3,4-thiadiazol-2-yl}benzenesulfonamide) was found to be the most selective inhibitor of the cancer-associated isoforms hCA IX and XII over the off-target isoforms, hCAI and II. The interaction dynamics and stability of <b>SUT16</b> within hCA IX and XII were investigated by molecular dynamics simulations as well as dynophore analysis. Based on computational data, increased hydrophobic contacts and hydrogen bonds in the tail part of these molecules within hCA IX and XII were found as favorable interactions leading to effective inhibitors of cancer-related isoforms.</p>","PeriodicalId":128,"journal":{"name":"Archiv der Pharmazie","volume":"357 11","pages":""},"PeriodicalIF":3.6000,"publicationDate":"2024-07-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ardp.202400439","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Archiv der Pharmazie","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/ardp.202400439","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
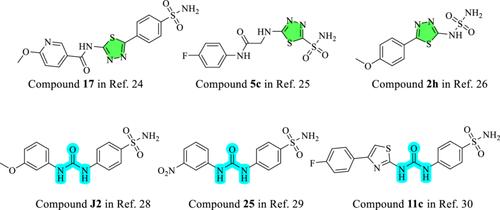
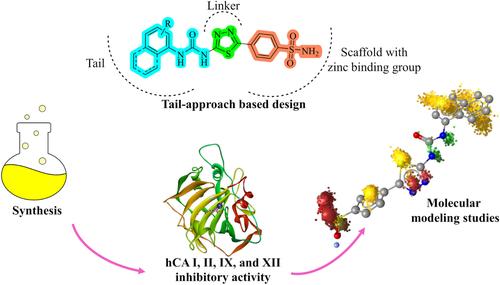
We synthesized herein 16 compounds (SUT1–SUT16) as potential carbonic anhydrase (CA) inhibitors utilizing the tail-approach design. Based on this strategy, we connected benzenesulfonamide, the zinc-binding scaffold, to different urea moieties with the 1,3,4-thiadiazole ring as a linker. We obtained the target compounds by the reaction of 4-(5-amino-1,3,4-thiadiazol-2-yl)benzenesulfonamide with aryl isocyanates. Upon confirmation of their structures, the compounds were screened for their ability to inhibit the tumor-related human (h) isoforms human carbonic anhydrase (hCA) IX and XII, as well as the physiologically dominant hCA I and II. Most of the molecules demonstrated Ki values ≤ 10 nM with different selectivity profiles. The binding modes of SUT9, SUT10, and SUT5, the most effective inhibitors of hCA II, IX, and XII, respectively, were predicted by molecular docking. SUT16 (4-{5-[3-(naphthalen-1-yl)ureido]-1,3,4-thiadiazol-2-yl}benzenesulfonamide) was found to be the most selective inhibitor of the cancer-associated isoforms hCA IX and XII over the off-target isoforms, hCAI and II. The interaction dynamics and stability of SUT16 within hCA IX and XII were investigated by molecular dynamics simulations as well as dynophore analysis. Based on computational data, increased hydrophobic contacts and hydrogen bonds in the tail part of these molecules within hCA IX and XII were found as favorable interactions leading to effective inhibitors of cancer-related isoforms.
我们利用尾部设计方法合成了 16 个化合物(SUT1-SUT16),作为潜在的碳酸酐酶(CA)抑制剂。根据这一策略,我们将锌结合支架苯磺酰胺与不同的脲基连接起来,并以 1,3,4-噻二唑环作为连接物。我们通过 4-(5-氨基-1,3,4-噻二唑-2-基)苯磺酰胺与芳基异氰酸酯的反应获得了目标化合物。在确认其结构后,对这些化合物进行了筛选,以检测其抑制与肿瘤相关的人类(h)碳酸酐酶(hCA)IX 和 XII 异构体以及生理上占主导地位的 hCA I 和 II 的能力。大多数分子的 Ki 值≤ 10 nM,具有不同的选择性。通过分子对接预测了 SUT9、SUT10 和 SUT5 的结合模式,它们分别是对 hCA II、IX 和 XII 最有效的抑制剂。研究发现,SUT16(4-{5-[3-(萘-1-基)脲基]-1,3,4-噻二唑-2-基}苯磺酰胺)是对癌症相关同工酶 hCA IX 和 XII 最具选择性的抑制剂,而非目标同工酶 hCAI 和 II。分子动力学模拟和动力学分析研究了 SUT16 在 hCA IX 和 XII 中的相互作用动力学和稳定性。根据计算数据,发现这些分子在 hCA IX 和 XII 中的尾部增加了疏水接触和氢键,这些有利的相互作用导致了对癌症相关同工酶的有效抑制。
期刊介绍:
Archiv der Pharmazie - Chemistry in Life Sciences is an international journal devoted to research and development in all fields of pharmaceutical and medicinal chemistry. Emphasis is put on papers combining synthetic organic chemistry, structural biology, molecular modelling, bioorganic chemistry, natural products chemistry, biochemistry or analytical methods with pharmaceutical or medicinal aspects such as biological activity. The focus of this journal is put on original research papers, but other scientifically valuable contributions (e.g. reviews, minireviews, highlights, symposia contributions, discussions, and essays) are also welcome.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
