Anna M. Aalbers, Paul L. A. van Daele, Virgil A. S. H. Dalm, Peter J. M. Valk, Marc H. G. P. Raaijmakers
{"title":"Long-term genetic and clinical remissions after cessation of azacitidine treatment in patients with VEXAS syndrome","authors":"Anna M. Aalbers, Paul L. A. van Daele, Virgil A. S. H. Dalm, Peter J. M. Valk, Marc H. G. P. Raaijmakers","doi":"10.1002/hem3.129","DOIUrl":null,"url":null,"abstract":"<p>VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) is an inflammatory syndrome caused by acquired mutations in the gene encoding ubiquitin like modifier activating enzyme 1 (<i>UBA1</i>) that is often fatal.<span><sup>1, 2</sup></span> Allogeneic hematopoietic stem cell transplantation is currently considered the only curative treatment modality.<span><sup>3-6</sup></span> We were the first to report eradication of virtually all <i>UBA1</i>-mutated cells by the hypomethylating agent azacitidine, reflected in clinical and genetic remissions,<span><sup>7</sup></span> a finding confirmed in a recent phase II clinical trial.<span><sup>8</sup></span></p><p>Here, we report persistent, long-term (11–84 months) genetic and clinical remissions in VEXAS patients responding to treatment with the hypomethylating agent azacitidine, after cessation of therapy. The data indicate that azacitidine treatment may be an attractive alternative to stem cell transplant for disease eradication in VEXAS syndrome patients and reveal long-term clonal stability of <i>UBA1</i>-mutated cells under homeostatic and inflammatory conditions.</p><p>Since its first description in December 2020,<span><sup>1</sup></span> a <i>UBA1</i> variant-confirmed diagnosis of VEXAS syndrome was made in 11 patients at our institution until February 2024 (all male, median age at diagnosis 67 years, range 57–77 years). <i>UBA1</i> mutation detection and panel-based sequencing in these patients was performed as previously reported<span><sup>7</sup></span> and as described in the Supporting Information Methods section. Of these 11 patients, eight have been exposed to azacitidine (administered at a dose of 75 mg/m<sup>2</sup> subcutaneously once daily for 7 days in a 4-weekly schedule). Of the three patients that were not exposed to azacitidine, two patients were treated with corticosteroids and deceased due to infectious complications, and one patient was considered not a candidate for azacitidine treatment due to psychosocial circumstances. In two patients, azacitidine was used as a last resort on an in-house basis, after failure of multiple other lines of treatment, at the time that patients were critically ill (WHO performance status 4) due to VEXAS-related (respiratory) pathology. Both patients died shortly after administration of the first cycle of azacitidine with clinically active disease, and before genetic assessment of response after the first cycle. Six patients received multiple cycles of azacitidine (range, 3–8 cycles) on an out-patient basis with genetic monitoring of disease response. The characteristics of these six patients are listed in Table 1. Patients 1 and 2 carried a concurrent <i>DNMT3A</i> mutation at diagnosis with a variant allele frequency (VAF) of 59% and 30%, respectively, and patient 3 carried a <i>TET2</i> mutation with a VAF of 4%. In patients 4, 5, and 6 no other mutations were detected by panel-based sequencing. Three of these six patients have been described by us before, including one patient in whom the diagnosis of VEXAS syndrome was made in retrospect, that is, after treatment with azacitidine.<span><sup>7</sup></span> Long-term follow-up of patients with an initial response to azacitidine is reported here. The study was performed in compliance with the Declaration of Helsinki, and reported patients gave informed consent for study participation.</p><p>All six VEXAS patients were treated with azacitidine because they suffered from frequent (life-threatening) inflammatory flares of disease and (in all but patients 4 and 5 transfusion-dependent) anemia, insufficiently responding to treatment with corticosteroids and disease-modifying antirheumatic drugs (DMARDs) and/or biologicals (specified in Table 1) and/or requiring long-term corticosteroid treatment with concomitant adverse events. Of note, four of six VEXAS patients formally fulfilled MDS criteria according to the WHO 2016 classification, based on the presence of cytopenia (anemia Hb <10 g/dL) and dysplasia (≥10% in any lineage), with clonality demonstrated by mutational analyses in three of these four patients.</p><p>Five (5/6) patients achieved a clinical and genetic response to azacitidine. In these patients, the VAF of the causative <i>UBA1</i> variant decreased from a median of 67% (range 56%–86%) in bone marrow or peripheral blood at start of treatment with azacitidine (indicating that the majority of cells in the bone marrow carried the mutation) to ≤1% in bone marrow and/or peripheral blood (range 0%–1%), which was documented after five cycles in four out of five patients (in patient 1, retrospective response assessment was performed firstly after eight cycles). <i>DNMT3A</i> VAF in patient 1 decreased from 59% to 1% and in patient 2 from 30% to 2%, consistent with the notion that azacitidine treatment resulted in the eradication of cells carrying both <i>DNMT3A</i> and <i>UBA1</i> variants in these patients. Genetic remissions, indicative of a dramatic reduction in the number of <i>UBA1</i>-mutated cells, were associated with complete clinical remissions, defined as complete absence of inflammatory flares of disease, as reflected by long-term normalization of C-reactive protein (CRP) in the blood plasma in these patients, and an increase in hemoglobin levels in all patients (Figure 1). All other immuno-modulatory drugs (e.g., corticosteroids, DMARDs and/or tocilizumab) could be weaned in all responding patients. In the three patients in whom a bone marrow evaluation was performed after achieving a clinical and genetic response, characteristic vacuolisation of erythroid and myeloid precursor cells, and dysplasia of erythroid, myeloid and/or megakaryocytic lineages, disappeared. The patient that was considered a nonresponder to azacitidine (patient 3) received a total of three cycles of azacitidine, after which the mutant <i>UBA1</i> VAF in bone marrow remained high (VAF 84%). <i>TET2</i> VAF remained similar before and after treatment (4% and 3%, respectively). Quality of life and anemia had not improved. Clinical symptoms of VEXAS were relatively mild at that time and treatment was stopped, which in retrospect might have been too early to achieve a genetic remission.</p><p>In the five responding patients, azacitidine treatment was stopped after achieving a genetic response, arbitrarily defined as mutant <i>UBA1</i> VAF < 5%. In patient 1 this was done after eight cycles because of treatment for a colon carcinoma, in two other patients the decision to interrupt/stop treatment upon achieving a response after four cycles was based on grade 2–3 adverse effects (fatigue and neutropenia) and the observation in patient 1 that genetic remissions may be maintained after drug cessation. The remaining two responding patients achieved a clinical and genetically-defined response (VAF of mutant <i>UBA1</i> from 75% to 0% in patient 5, and from 56% to 0% in patient 6) after five cycles recently (at submission of this manuscript, May 2024) and treatment with azacitidine was discontinued after this fifth cycle based on the clinical and genetic response and previous experience with cessation of azacitidine in the other patients. All other immunomodulatory treatment (as specified in Table 1) could be completely stopped in all responding patients, except for patient 1 who continues on prednisolone 5 mg daily for VEXAS-unrelated reasons.</p><p>After cessation of azacitidine treatment, the first three responding patients (in whom follow-up after cessation of treatment is present) remained in a clinical and genetic remission with a current median follow up of 31 months (range 11–84 months) after the last cycle of azacitidine (Figure 1). Patients remain free of any VEXAS-related inflammatory manifestations and maintain stable normalized blood levels. The mutant <i>UBA1</i> VAF in the blood remained relatively stable (range 0%–7%) in all patients during this median follow up of multiple years. We did observe a very gradual increase of the mutant <i>UBA1</i> VAF (from 1% to 7%) in patient 1 over the course of 7 years. Of further interest, mutant <i>UBA1</i> VAF was not notably affected by inflammatory episodes caused by a Campylobacter jejuni PCR-positive gastro-enteritis requiring hospitalization (patient 1) and an episode of crystal proven gout requiring hospitalization (patient 2) (Figure 1).</p><p>Collectively, these findings confirm relatively high rates of response to azacitidine treatment in VEXAS syndrome, as previously reported by us and others,<span><sup>7-10</sup></span> and, more importantly, indicate that azacitidine may be safely stopped upon achievement of a genetic response, resulting in long-term genetic and clinical remissions. This is of significant clinical relevance because it would position azacitidine treatment as an attractive alternative to stem cell transplant as the only currently available long-term disease eradicating therapeutic approach in VEXAS syndrome patients. Furthermore, cessation of azacitidine treatment would safeguard patients from frequently occurring adverse effects of the drug, including fatigue, myelosuppression and infectious complications, affecting quality of life. The data warrant incorporation of drug interruption (“holiday”) designs in future prospective clinical trials investigating the value of azacitidine treatment for VEXAS syndrome. The mechanism by which azacitidine eradicates <i>UBA1</i>-mutated cells is not known to date, but it may be hypothesized that a defect in the ubiquitin-proteasome system sensitizes cells to this agent.</p><p>Finally, findings might shed new light on the biology of disease and long-term kinetics of mutant <i>UBA1</i> clones, demonstrating that they can remain stable for many years, even in elderly patients. Although the number of documented infectious or inflammatory episodes is too small to draw conclusions, the data suggest that mutant <i>UBA1</i> clones may remain stable not only under “homeostatic” conditions but also in the event of “inflammatory stress” caused by infectious or inflammatory conditions. It may be speculated that clonal evolution of <i>UBA1</i> mutant cells takes many years and, ultimately, clones may be maintained by mutant <i>UBA1</i>-driven bone marrow inflammation, above a critical threshold, in a feed-forward manner. Breaking this feed-forward loop (with azacitidine) may thus result in long-term genetic and clinical remission in these patients, a notion that awaits experimental support and/or further long-term observation of VEXAS patients.</p><p>Marc H. G. P. Raaijmakers conceived and supervised the study. Marc H. G. P. Raaijmakers and Anna M. Aalbers were responsible for the clinical care of patients, collected data, and wrote the manuscript. Paul L. A. van Daele and Virgil A. S. H. Dalm were responsible for the clinical care of patients, collected data and edited the manuscript. Peter J. M. Valk coordinated genetic analyses and edited the manuscript.</p><p>The authors declare no conflict of interest.</p><p>This research received no funding.</p>","PeriodicalId":12982,"journal":{"name":"HemaSphere","volume":"8 8","pages":""},"PeriodicalIF":14.6000,"publicationDate":"2024-07-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11287193/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"HemaSphere","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/hem3.129","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"HEMATOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) is an inflammatory syndrome caused by acquired mutations in the gene encoding ubiquitin like modifier activating enzyme 1 (UBA1) that is often fatal.1, 2 Allogeneic hematopoietic stem cell transplantation is currently considered the only curative treatment modality.3-6 We were the first to report eradication of virtually all UBA1-mutated cells by the hypomethylating agent azacitidine, reflected in clinical and genetic remissions,7 a finding confirmed in a recent phase II clinical trial.8
Here, we report persistent, long-term (11–84 months) genetic and clinical remissions in VEXAS patients responding to treatment with the hypomethylating agent azacitidine, after cessation of therapy. The data indicate that azacitidine treatment may be an attractive alternative to stem cell transplant for disease eradication in VEXAS syndrome patients and reveal long-term clonal stability of UBA1-mutated cells under homeostatic and inflammatory conditions.
Since its first description in December 2020,1 a UBA1 variant-confirmed diagnosis of VEXAS syndrome was made in 11 patients at our institution until February 2024 (all male, median age at diagnosis 67 years, range 57–77 years). UBA1 mutation detection and panel-based sequencing in these patients was performed as previously reported7 and as described in the Supporting Information Methods section. Of these 11 patients, eight have been exposed to azacitidine (administered at a dose of 75 mg/m2 subcutaneously once daily for 7 days in a 4-weekly schedule). Of the three patients that were not exposed to azacitidine, two patients were treated with corticosteroids and deceased due to infectious complications, and one patient was considered not a candidate for azacitidine treatment due to psychosocial circumstances. In two patients, azacitidine was used as a last resort on an in-house basis, after failure of multiple other lines of treatment, at the time that patients were critically ill (WHO performance status 4) due to VEXAS-related (respiratory) pathology. Both patients died shortly after administration of the first cycle of azacitidine with clinically active disease, and before genetic assessment of response after the first cycle. Six patients received multiple cycles of azacitidine (range, 3–8 cycles) on an out-patient basis with genetic monitoring of disease response. The characteristics of these six patients are listed in Table 1. Patients 1 and 2 carried a concurrent DNMT3A mutation at diagnosis with a variant allele frequency (VAF) of 59% and 30%, respectively, and patient 3 carried a TET2 mutation with a VAF of 4%. In patients 4, 5, and 6 no other mutations were detected by panel-based sequencing. Three of these six patients have been described by us before, including one patient in whom the diagnosis of VEXAS syndrome was made in retrospect, that is, after treatment with azacitidine.7 Long-term follow-up of patients with an initial response to azacitidine is reported here. The study was performed in compliance with the Declaration of Helsinki, and reported patients gave informed consent for study participation.
All six VEXAS patients were treated with azacitidine because they suffered from frequent (life-threatening) inflammatory flares of disease and (in all but patients 4 and 5 transfusion-dependent) anemia, insufficiently responding to treatment with corticosteroids and disease-modifying antirheumatic drugs (DMARDs) and/or biologicals (specified in Table 1) and/or requiring long-term corticosteroid treatment with concomitant adverse events. Of note, four of six VEXAS patients formally fulfilled MDS criteria according to the WHO 2016 classification, based on the presence of cytopenia (anemia Hb <10 g/dL) and dysplasia (≥10% in any lineage), with clonality demonstrated by mutational analyses in three of these four patients.
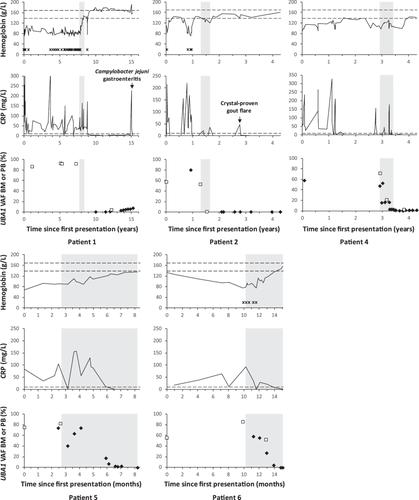
Five (5/6) patients achieved a clinical and genetic response to azacitidine. In these patients, the VAF of the causative UBA1 variant decreased from a median of 67% (range 56%–86%) in bone marrow or peripheral blood at start of treatment with azacitidine (indicating that the majority of cells in the bone marrow carried the mutation) to ≤1% in bone marrow and/or peripheral blood (range 0%–1%), which was documented after five cycles in four out of five patients (in patient 1, retrospective response assessment was performed firstly after eight cycles). DNMT3A VAF in patient 1 decreased from 59% to 1% and in patient 2 from 30% to 2%, consistent with the notion that azacitidine treatment resulted in the eradication of cells carrying both DNMT3A and UBA1 variants in these patients. Genetic remissions, indicative of a dramatic reduction in the number of UBA1-mutated cells, were associated with complete clinical remissions, defined as complete absence of inflammatory flares of disease, as reflected by long-term normalization of C-reactive protein (CRP) in the blood plasma in these patients, and an increase in hemoglobin levels in all patients (Figure 1). All other immuno-modulatory drugs (e.g., corticosteroids, DMARDs and/or tocilizumab) could be weaned in all responding patients. In the three patients in whom a bone marrow evaluation was performed after achieving a clinical and genetic response, characteristic vacuolisation of erythroid and myeloid precursor cells, and dysplasia of erythroid, myeloid and/or megakaryocytic lineages, disappeared. The patient that was considered a nonresponder to azacitidine (patient 3) received a total of three cycles of azacitidine, after which the mutant UBA1 VAF in bone marrow remained high (VAF 84%). TET2 VAF remained similar before and after treatment (4% and 3%, respectively). Quality of life and anemia had not improved. Clinical symptoms of VEXAS were relatively mild at that time and treatment was stopped, which in retrospect might have been too early to achieve a genetic remission.
In the five responding patients, azacitidine treatment was stopped after achieving a genetic response, arbitrarily defined as mutant UBA1 VAF < 5%. In patient 1 this was done after eight cycles because of treatment for a colon carcinoma, in two other patients the decision to interrupt/stop treatment upon achieving a response after four cycles was based on grade 2–3 adverse effects (fatigue and neutropenia) and the observation in patient 1 that genetic remissions may be maintained after drug cessation. The remaining two responding patients achieved a clinical and genetically-defined response (VAF of mutant UBA1 from 75% to 0% in patient 5, and from 56% to 0% in patient 6) after five cycles recently (at submission of this manuscript, May 2024) and treatment with azacitidine was discontinued after this fifth cycle based on the clinical and genetic response and previous experience with cessation of azacitidine in the other patients. All other immunomodulatory treatment (as specified in Table 1) could be completely stopped in all responding patients, except for patient 1 who continues on prednisolone 5 mg daily for VEXAS-unrelated reasons.
After cessation of azacitidine treatment, the first three responding patients (in whom follow-up after cessation of treatment is present) remained in a clinical and genetic remission with a current median follow up of 31 months (range 11–84 months) after the last cycle of azacitidine (Figure 1). Patients remain free of any VEXAS-related inflammatory manifestations and maintain stable normalized blood levels. The mutant UBA1 VAF in the blood remained relatively stable (range 0%–7%) in all patients during this median follow up of multiple years. We did observe a very gradual increase of the mutant UBA1 VAF (from 1% to 7%) in patient 1 over the course of 7 years. Of further interest, mutant UBA1 VAF was not notably affected by inflammatory episodes caused by a Campylobacter jejuni PCR-positive gastro-enteritis requiring hospitalization (patient 1) and an episode of crystal proven gout requiring hospitalization (patient 2) (Figure 1).
Collectively, these findings confirm relatively high rates of response to azacitidine treatment in VEXAS syndrome, as previously reported by us and others,7-10 and, more importantly, indicate that azacitidine may be safely stopped upon achievement of a genetic response, resulting in long-term genetic and clinical remissions. This is of significant clinical relevance because it would position azacitidine treatment as an attractive alternative to stem cell transplant as the only currently available long-term disease eradicating therapeutic approach in VEXAS syndrome patients. Furthermore, cessation of azacitidine treatment would safeguard patients from frequently occurring adverse effects of the drug, including fatigue, myelosuppression and infectious complications, affecting quality of life. The data warrant incorporation of drug interruption (“holiday”) designs in future prospective clinical trials investigating the value of azacitidine treatment for VEXAS syndrome. The mechanism by which azacitidine eradicates UBA1-mutated cells is not known to date, but it may be hypothesized that a defect in the ubiquitin-proteasome system sensitizes cells to this agent.
Finally, findings might shed new light on the biology of disease and long-term kinetics of mutant UBA1 clones, demonstrating that they can remain stable for many years, even in elderly patients. Although the number of documented infectious or inflammatory episodes is too small to draw conclusions, the data suggest that mutant UBA1 clones may remain stable not only under “homeostatic” conditions but also in the event of “inflammatory stress” caused by infectious or inflammatory conditions. It may be speculated that clonal evolution of UBA1 mutant cells takes many years and, ultimately, clones may be maintained by mutant UBA1-driven bone marrow inflammation, above a critical threshold, in a feed-forward manner. Breaking this feed-forward loop (with azacitidine) may thus result in long-term genetic and clinical remission in these patients, a notion that awaits experimental support and/or further long-term observation of VEXAS patients.
Marc H. G. P. Raaijmakers conceived and supervised the study. Marc H. G. P. Raaijmakers and Anna M. Aalbers were responsible for the clinical care of patients, collected data, and wrote the manuscript. Paul L. A. van Daele and Virgil A. S. H. Dalm were responsible for the clinical care of patients, collected data and edited the manuscript. Peter J. M. Valk coordinated genetic analyses and edited the manuscript.
期刊介绍:
HemaSphere, as a publication, is dedicated to disseminating the outcomes of profoundly pertinent basic, translational, and clinical research endeavors within the field of hematology. The journal actively seeks robust studies that unveil novel discoveries with significant ramifications for hematology.
In addition to original research, HemaSphere features review articles and guideline articles that furnish lucid synopses and discussions of emerging developments, along with recommendations for patient care.
Positioned as the foremost resource in hematology, HemaSphere augments its offerings with specialized sections like HemaTopics and HemaPolicy. These segments engender insightful dialogues covering a spectrum of hematology-related topics, including digestible summaries of pivotal articles, updates on new therapies, deliberations on European policy matters, and other noteworthy news items within the field. Steering the course of HemaSphere are Editor in Chief Jan Cools and Deputy Editor in Chief Claire Harrison, alongside the guidance of an esteemed Editorial Board comprising international luminaries in both research and clinical realms, each representing diverse areas of hematologic expertise.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
