Justin R. Randall, Luiz C. Vieira, Claus O. Wilke, Bryan W. Davies
{"title":"Deep mutational scanning and machine learning for the analysis of antimicrobial-peptide features driving membrane selectivity","authors":"Justin R. Randall, Luiz C. Vieira, Claus O. Wilke, Bryan W. Davies","doi":"10.1038/s41551-024-01243-1","DOIUrl":null,"url":null,"abstract":"Many antimicrobial peptides directly disrupt bacterial membranes yet can also damage mammalian membranes. It is therefore central to their therapeutic use that rules governing the membrane selectivity of antimicrobial peptides be deciphered. However, this is difficult even for short peptides owing to the large combinatorial space of amino acid sequences. Here we describe a method for measuring the loss or maintenance of antimicrobial-peptide activity for thousands of peptide-sequence variants simultaneously, and its application to Protegrin-1, a potent yet toxic antimicrobial peptide, to determine the positional importance and flexibility of residues across its sequence while identifying variants with changes in membrane selectivity. More bacterially selective variants maintained a membrane-bound secondary structure while avoiding aromatic residues and cysteine pairs. A machine-learning model trained with our datasets accurately predicted membrane-specific activities for over 5.7 million Protegrin-1 variants, and identified one variant that showed substantially reduced toxicity and retention of activity in a mouse model of intraperitoneal infection. The high-throughput methodology may help elucidate sequence–structure–function relationships in antimicrobial peptides and inform the design of peptide-based synthetic drugs. A high-throughput mutation-scanning method for interrogating sequence variation in antimicrobial peptides facilitates the understanding of sequence–structure–function relationships affecting the membrane selectivity of antimicrobial peptides.","PeriodicalId":19063,"journal":{"name":"Nature Biomedical Engineering","volume":"8 7","pages":"842-853"},"PeriodicalIF":26.8000,"publicationDate":"2024-07-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature Biomedical Engineering","FirstCategoryId":"5","ListUrlMain":"https://www.nature.com/articles/s41551-024-01243-1","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ENGINEERING, BIOMEDICAL","Score":null,"Total":0}
引用次数: 0
Abstract
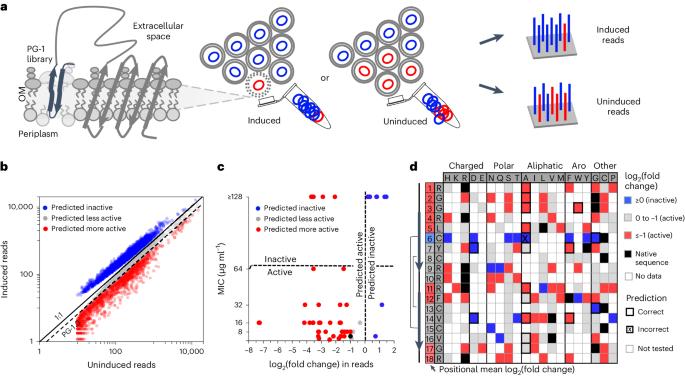
Many antimicrobial peptides directly disrupt bacterial membranes yet can also damage mammalian membranes. It is therefore central to their therapeutic use that rules governing the membrane selectivity of antimicrobial peptides be deciphered. However, this is difficult even for short peptides owing to the large combinatorial space of amino acid sequences. Here we describe a method for measuring the loss or maintenance of antimicrobial-peptide activity for thousands of peptide-sequence variants simultaneously, and its application to Protegrin-1, a potent yet toxic antimicrobial peptide, to determine the positional importance and flexibility of residues across its sequence while identifying variants with changes in membrane selectivity. More bacterially selective variants maintained a membrane-bound secondary structure while avoiding aromatic residues and cysteine pairs. A machine-learning model trained with our datasets accurately predicted membrane-specific activities for over 5.7 million Protegrin-1 variants, and identified one variant that showed substantially reduced toxicity and retention of activity in a mouse model of intraperitoneal infection. The high-throughput methodology may help elucidate sequence–structure–function relationships in antimicrobial peptides and inform the design of peptide-based synthetic drugs. A high-throughput mutation-scanning method for interrogating sequence variation in antimicrobial peptides facilitates the understanding of sequence–structure–function relationships affecting the membrane selectivity of antimicrobial peptides.
期刊介绍:
Nature Biomedical Engineering is an online-only monthly journal that was launched in January 2017. It aims to publish original research, reviews, and commentary focusing on applied biomedicine and health technology. The journal targets a diverse audience, including life scientists who are involved in developing experimental or computational systems and methods to enhance our understanding of human physiology. It also covers biomedical researchers and engineers who are engaged in designing or optimizing therapies, assays, devices, or procedures for diagnosing or treating diseases. Additionally, clinicians, who make use of research outputs to evaluate patient health or administer therapy in various clinical settings and healthcare contexts, are also part of the target audience.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
