{"title":"Machine-learned interatomic potentials for transition metal dichalcogenide Mo1−xWxS2−2ySe2y alloys","authors":"Anas Siddiqui, Nicholas D. M. Hine","doi":"10.1038/s41524-024-01357-9","DOIUrl":null,"url":null,"abstract":"<p>Machine Learned Interatomic Potentials (MLIPs) combine the predictive power of Density Functional Theory (DFT) with the speed and scaling of interatomic potentials, enabling theoretical spectroscopy to be applied to larger and more complex systems than is possible with DFT. In this work, we train an MLIP for quaternary Transition Metal Dichalcogenide (TMD) alloy systems of the form Mo<sub>1−<i>x</i></sub>W<sub><i>x</i></sub>S<sub>2−2<i>y</i></sub>Se<sub>2<i>y</i></sub>, using the equivariant Neural Network (NN) MACE. We demonstrate the ability of this potential to calculate vibrational properties of alloy TMDs including phonon spectra for pure monolayers, and Vibrational Density of States (VDOS) and first-order Raman spectra for alloys across the range of <i>x</i> and <i>y</i>. We show that we retain DFT level accuracy while greatly extending feasible system size and extent of sampling over alloy configurations. We are able to characterize the first-order Raman active modes across the whole range of concentration, particularly for the “disorder-induced” modes.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"41 1","pages":""},"PeriodicalIF":11.9000,"publicationDate":"2024-08-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-024-01357-9","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
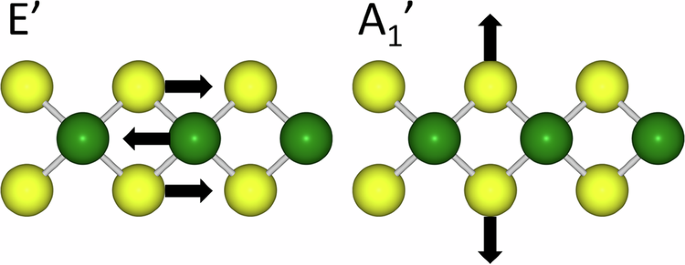
Machine Learned Interatomic Potentials (MLIPs) combine the predictive power of Density Functional Theory (DFT) with the speed and scaling of interatomic potentials, enabling theoretical spectroscopy to be applied to larger and more complex systems than is possible with DFT. In this work, we train an MLIP for quaternary Transition Metal Dichalcogenide (TMD) alloy systems of the form Mo1−xWxS2−2ySe2y, using the equivariant Neural Network (NN) MACE. We demonstrate the ability of this potential to calculate vibrational properties of alloy TMDs including phonon spectra for pure monolayers, and Vibrational Density of States (VDOS) and first-order Raman spectra for alloys across the range of x and y. We show that we retain DFT level accuracy while greatly extending feasible system size and extent of sampling over alloy configurations. We are able to characterize the first-order Raman active modes across the whole range of concentration, particularly for the “disorder-induced” modes.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
