Unveiling the Structure and Dynamics of Ac3+ Ion in Aqueous Solution: Insight From Relativistic Hybrid Forces Molecular Mechanics Molecular Dynamics Simulations
Muhammad Aditya Abimanyu, Niko Prasetyo, Mokhammad Fajar Pradipta
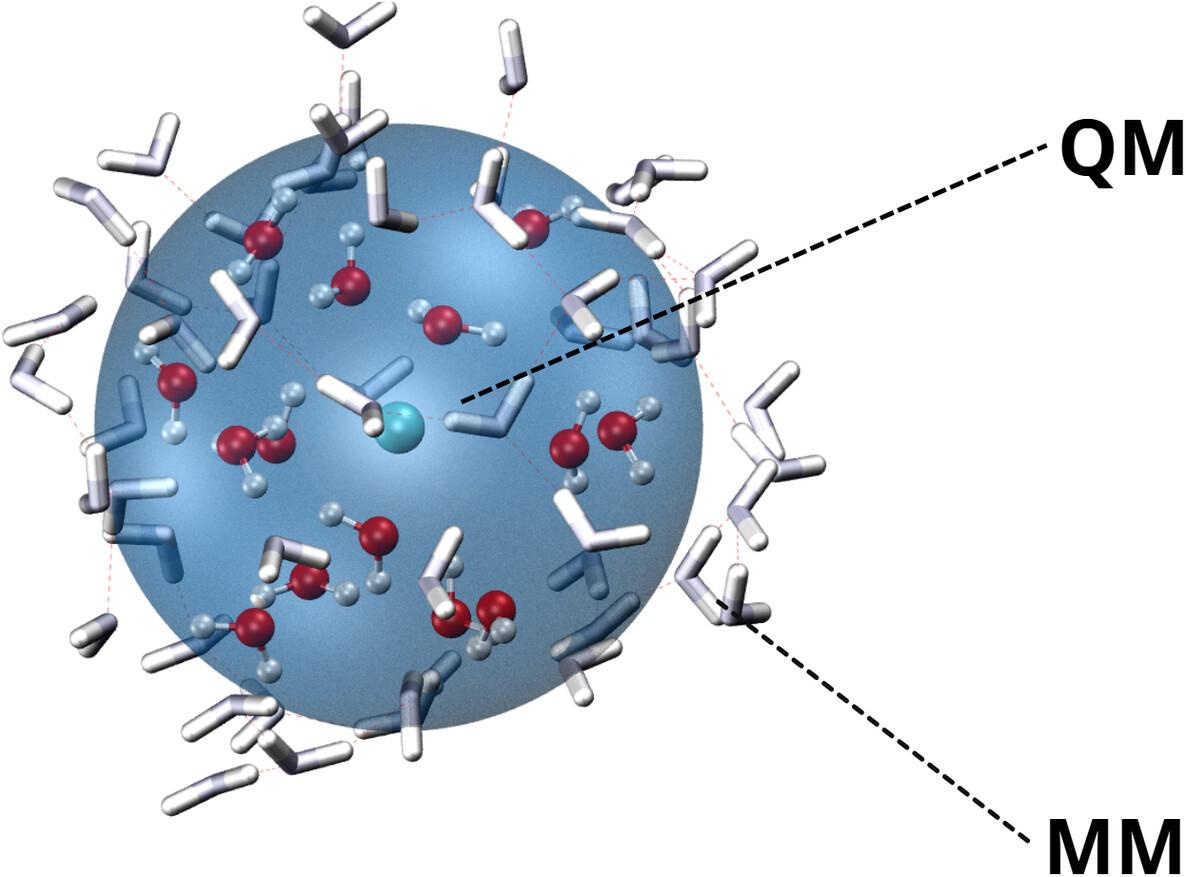
{"title":"Unveiling the Structure and Dynamics of Ac3+ Ion in Aqueous Solution: Insight From Relativistic Hybrid Forces Molecular Mechanics Molecular Dynamics Simulations","authors":"Muhammad Aditya Abimanyu, Niko Prasetyo, Mokhammad Fajar Pradipta","doi":"10.1002/qua.27464","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>This work describes a molecular dynamics simulation study (MP2-DKH2/MM) that explores the structural and dynamical properties of hydrated Ac<sup>3+</sup> ions in an aqueous solution. Simulation results indicate that the ion formed three hydration shells. The hydrated Ac<sup>3+</sup> had a first hydration shell comprising 8–9 water molecules. It showed similar probabilities for both coordination numbers, showing a flexible first hydration shell with eight registered successful ligand exchanges during the simulation. The water molecules' mean residence times (MRT) in the first, second, and third hydration shells were 131.8, 6.46, and 2.67 ps, respectively. The complexes of octahydrate ([Ac(H₂O)₈]<sup>3+</sup>) and nonahydrate ([Ac(H₂O)₉]<sup>3+</sup>) were observed in the first hydration shell. The square antiprism (SA) geometry was adopted for octahydrate, while the gyroelongated square antiprism (GySA) geometry was adopted for nonahydrate. The simulations provided valuable insights into the ion-oxygen stretching frequencies. Specifically, the average stretching frequency for Ac<sup>3+</sup> was found to be 404 cm<sup>−1</sup>, which is in good agreement with the calculated value from the CCSD(T) calculation of 398.78 cm<sup>−1</sup>. These findings indicate that including DKH2 relativistic approximation increases the accuracy of the simulation results and can contribute to understanding these actinide ions' behavior in aqueous environments, shedding light on hydrated systems' structural arrangements and dynamics.</p>\n </div>","PeriodicalId":182,"journal":{"name":"International Journal of Quantum Chemistry","volume":"124 16","pages":""},"PeriodicalIF":2.0000,"publicationDate":"2024-08-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Quantum Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/qua.27464","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
This work describes a molecular dynamics simulation study (MP2-DKH2/MM) that explores the structural and dynamical properties of hydrated Ac3+ ions in an aqueous solution. Simulation results indicate that the ion formed three hydration shells. The hydrated Ac3+ had a first hydration shell comprising 8–9 water molecules. It showed similar probabilities for both coordination numbers, showing a flexible first hydration shell with eight registered successful ligand exchanges during the simulation. The water molecules' mean residence times (MRT) in the first, second, and third hydration shells were 131.8, 6.46, and 2.67 ps, respectively. The complexes of octahydrate ([Ac(H₂O)₈]3+) and nonahydrate ([Ac(H₂O)₉]3+) were observed in the first hydration shell. The square antiprism (SA) geometry was adopted for octahydrate, while the gyroelongated square antiprism (GySA) geometry was adopted for nonahydrate. The simulations provided valuable insights into the ion-oxygen stretching frequencies. Specifically, the average stretching frequency for Ac3+ was found to be 404 cm−1, which is in good agreement with the calculated value from the CCSD(T) calculation of 398.78 cm−1. These findings indicate that including DKH2 relativistic approximation increases the accuracy of the simulation results and can contribute to understanding these actinide ions' behavior in aqueous environments, shedding light on hydrated systems' structural arrangements and dynamics.
期刊介绍:
Since its first formulation quantum chemistry has provided the conceptual and terminological framework necessary to understand atoms, molecules and the condensed matter. Over the past decades synergistic advances in the methodological developments, software and hardware have transformed quantum chemistry in a truly interdisciplinary science that has expanded beyond its traditional core of molecular sciences to fields as diverse as chemistry and catalysis, biophysics, nanotechnology and material science.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
