Banan O. Alomari, Lara I. Fakhouri, Nizar A. Al‑Shar’i, Qosay Albalas
{"title":"Design, synthesis and biological evaluation of potent thiazolidinedione salicylic acid inhibitors against glyoxalase-I as potential anticancer agents","authors":"Banan O. Alomari, Lara I. Fakhouri, Nizar A. Al‑Shar’i, Qosay Albalas","doi":"10.1007/s00044-024-03247-7","DOIUrl":null,"url":null,"abstract":"<div><p>The worldwide rise in cancer incidence and mortality rates has spurred the search for new pathways implicated in cancer development and progression. One such target is glyoxalase 1 (GLO-I), a key player in methylglyoxal detoxification and a factor in the proliferation and prognosis of numerous cancers. Recent studies led by Al-Shar’i et al. utilized computer-aided drug design to identify potential inhibitors of GLO-I. The second most potent hit, (<i>Z</i>)-5-(5-((2,4-dioxothiazolidin-5-ylidene)methyl)furan-2-yl)-2-hydroxybenzoic acid, (IC<sub><i>50</i></sub> = 4.24 µM), was selected as a lead for further optimization. Through molecular docking, 27 analogs were designed and evaluated for binding affinity, with 14 of the top-scorings synthesized and tested for their inhibitory activity against GLO-I. The majority of these analogs showed enhanced activities relative to the lead compound, with the most potent having an IC<sub>50</sub> of 150 nM. These findings pave the way for the continued development of highly effective GLO-I inhibitors.</p><div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>","PeriodicalId":699,"journal":{"name":"Medicinal Chemistry Research","volume":"33 9","pages":"1526 - 1540"},"PeriodicalIF":3.1000,"publicationDate":"2024-08-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Medicinal Chemistry Research","FirstCategoryId":"3","ListUrlMain":"https://link.springer.com/article/10.1007/s00044-024-03247-7","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
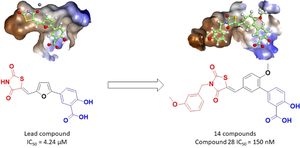
The worldwide rise in cancer incidence and mortality rates has spurred the search for new pathways implicated in cancer development and progression. One such target is glyoxalase 1 (GLO-I), a key player in methylglyoxal detoxification and a factor in the proliferation and prognosis of numerous cancers. Recent studies led by Al-Shar’i et al. utilized computer-aided drug design to identify potential inhibitors of GLO-I. The second most potent hit, (Z)-5-(5-((2,4-dioxothiazolidin-5-ylidene)methyl)furan-2-yl)-2-hydroxybenzoic acid, (IC50 = 4.24 µM), was selected as a lead for further optimization. Through molecular docking, 27 analogs were designed and evaluated for binding affinity, with 14 of the top-scorings synthesized and tested for their inhibitory activity against GLO-I. The majority of these analogs showed enhanced activities relative to the lead compound, with the most potent having an IC50 of 150 nM. These findings pave the way for the continued development of highly effective GLO-I inhibitors.
期刊介绍:
Medicinal Chemistry Research (MCRE) publishes papers on a wide range of topics, favoring research with significant, new, and up-to-date information. Although the journal has a demanding peer review process, MCRE still boasts rapid publication, due in part, to the length of the submissions. The journal publishes significant research on various topics, many of which emphasize the structure-activity relationships of molecular biology.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
