{"title":"Enhanced thermoelectric performance of Hf-doped ZrNiSn: a first principle study","authors":"Di Cao, Jiannong Cao","doi":"10.1007/s00894-024-06102-z","DOIUrl":null,"url":null,"abstract":"<div><h3>Context and results</h3><p>In this work, we perform a systematic study on the thermoelectric properties of Zr<sub>1-x</sub>NiSnHf<sub>x</sub> using first-principles calculations combined with Boltzmann transport equations. The power factor of Zr<sub>1-x</sub>NiSnHf<sub>x</sub> increases as the temperature increases from 300 to 1200 K, because the increase in electrical conductivity is greater than the decrease in the Seebeck coefficient. The power factor of Zr<sub>7/8</sub>NiSnHf<sub>1/8</sub> is larger than that of other Zr<sub>1-x</sub>NiSnHf<sub>x</sub> thermoelectric materials, but the thermoelectric figure of merit (ZT) is similar to that of others materials. This is due to the higher electronic thermal conductivity of Zr<sub>7/8</sub>NiSnHf<sub>1/8</sub> compared to other materials. The maximum ZT of p-type (n-type) Zr<sub>1-x</sub>NiSnHf<sub>x</sub> is 0.98 (0.97), 0.9 (0.89), 0.83 (0.80), and 0.72 (0.73) at 300 K, 600 K, 900 K, and 1200 K, respectively, which are greater than those of the pure ZrNiSn. In conclusion, Hf-doped ZrNiSn can enhance the thermoelectric performance and are promising candidates for thermoelectric materials.</p><h3>Computational method</h3><p>This paper uses FP-LAPW implemented in the WIEN2K code. The thermoelectric performance is calculated based on the semi-classical Boltzmann theory implanted using the BoltzTraP code. The electronic thermal conductivity (<i>κ</i><sub>e</sub>) and the carrier concentration (<i>n</i>) have been calculated using the density functional theory.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"30 9","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2024-08-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-024-06102-z","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Context and results
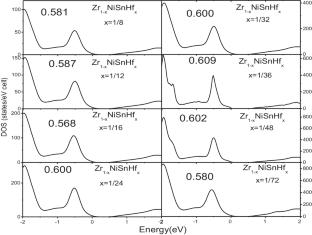
In this work, we perform a systematic study on the thermoelectric properties of Zr1-xNiSnHfx using first-principles calculations combined with Boltzmann transport equations. The power factor of Zr1-xNiSnHfx increases as the temperature increases from 300 to 1200 K, because the increase in electrical conductivity is greater than the decrease in the Seebeck coefficient. The power factor of Zr7/8NiSnHf1/8 is larger than that of other Zr1-xNiSnHfx thermoelectric materials, but the thermoelectric figure of merit (ZT) is similar to that of others materials. This is due to the higher electronic thermal conductivity of Zr7/8NiSnHf1/8 compared to other materials. The maximum ZT of p-type (n-type) Zr1-xNiSnHfx is 0.98 (0.97), 0.9 (0.89), 0.83 (0.80), and 0.72 (0.73) at 300 K, 600 K, 900 K, and 1200 K, respectively, which are greater than those of the pure ZrNiSn. In conclusion, Hf-doped ZrNiSn can enhance the thermoelectric performance and are promising candidates for thermoelectric materials.
Computational method
This paper uses FP-LAPW implemented in the WIEN2K code. The thermoelectric performance is calculated based on the semi-classical Boltzmann theory implanted using the BoltzTraP code. The electronic thermal conductivity (κe) and the carrier concentration (n) have been calculated using the density functional theory.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
