{"title":"Ancestral duplication of MADS-box genes in land plants empowered the functional divergence between sporophytes and gametophytes","authors":"Yichun Qiu, Zhen Li, Claudia Köhler","doi":"10.1111/nph.20065","DOIUrl":null,"url":null,"abstract":"<p>MADS-box transcription factors (TFs) have gained widespread recognition for their exceptional diversity and pivotal roles in various biological functions across eukaryotic organisms. Specifically, in land plants, the MADS-box gene family has undergone substantial expansion and provided the genetic raw material for many developmental novelties, including flowers, fruits and seeds. Therefore, understanding the origin of MADS-box genes is crucial for gaining insights into the evolutionary success of land plants. Land plant MADS-box TFs have been categorized into two groups. MIKC-type (Type II) TFs are named after the structural arrangement of the MADS (M), Intervening (I) and plant-specific Keratin-like (K) domain, followed by a variable C-terminal region. By contrast, M-type (Type I) TFs lack the K domain (Alvarez-Buylla <i>et al</i>., <span>2000</span>; Kofuji <i>et al</i>., <span>2003</span>; Nam <i>et al</i>., <span>2004</span>). The identification of mutants exhibiting distinct flower organ patterns led to the discovery of a multitude of MIKC-type MADS-box TFs in plants (Ng & Yanofsky, <span>2001</span>; Nam <i>et al</i>., <span>2003</span>; Kaufmann <i>et al</i>., <span>2005</span>). By contrast, the identification of M-type genes occurred exclusively through bioinformatic analyses following the unveiling of the <i>Arabidopsis thaliana</i> genome (Alvarez-Buylla <i>et al</i>., <span>2000</span>). Within the MIKC-type of MADS-box TFs, the MIKC*-type is distinguished based on the different arrangements of exons that encode the K domain (Svensson <i>et al</i>., <span>2000</span>; Henschel <i>et al</i>., <span>2002</span>; Zobell <i>et al</i>., <span>2010</span>; Kwantes <i>et al</i>., <span>2012</span>; Rümpler <i>et al</i>., <span>2023</span>). Both the ‘classic’ MIKC<sup>C</sup>- and the MIKC*-types are present across land plant lineages and form well-supported separate clades (Kwantes <i>et al</i>., <span>2012</span>; Gramzow & Theissen, <span>2013</span>; Liu <i>et al</i>., <span>2013</span>), suggesting that they diverged before the diversification of land plant lineages (Henschel <i>et al</i>., <span>2002</span>; Kofuji <i>et al</i>., <span>2003</span>; Tanabe <i>et al</i>., <span>2005</span>; Kwantes <i>et al</i>., <span>2012</span>).</p><p>It was a long-standing prevailing view in the field that plant Type II genes are orthologs to myocyte enhancer factor-2 (MEF2) genes in animals. Those were presumed to have diverged from plant Type I genes and animal serum response factor (SRF) genes via an ancient duplication before the divergence of the extant eukaryotic lineages (Alvarez-Buylla <i>et al</i>., <span>2000</span>). Thus, with the shared presence of the K domain, MIKC*-type MADS-box genes were intuitively considered more closely related to MIKC<sup>C</sup>-type genes, collectively called Type II. This adheres to the principle of parsimony, by which the acquisition of the K domain should ideally have occurred only once during the evolution of plants (Kaufmann <i>et al</i>., <span>2005</span>; Thangavel & Nayar, <span>2018</span>) (Fig. 1a). However, we recently showed that both Type I and Type II genes are land plant-specific MEF2 orthologs that arose from a duplication event predating the origin of land plants (Qiu <i>et al</i>., <span>2023</span>). Therefore, the plant Type I and Type II genes are more closely related than previously appreciated (Fig. 1a). Noticeably, the Type I and II gene duplication hypothetically took place around the time inferred for the rise of MIKC*-type genes (Kofuji <i>et al</i>., <span>2003</span>; Kwantes <i>et al</i>., <span>2012</span>). This raises the question whether the previously proposed monophyletic relationship between the MIKC*- and MIKC<sup>C</sup>-type as the Type II clade is better supported than the alternative hypothesis proposing that MIKC*-type genes are close paralogs of the Type I (M-type) clade (Fig. 1a). Since the ancestral land plant MADS-box gene giving rise to all current subfamilies has been inferred to include the K domain (Kaufmann <i>et al</i>., <span>2005</span>; Thangavel & Nayar, <span>2018</span>; Qiu <i>et al</i>., <span>2023</span>), this domain is not only shared by MIKC<sup>C</sup>-type and MIKC*-type, but also inherited by the ancestral gene leading to the extant Type I clade. Therefore, MIKC<sup>C</sup>- and MIKC*-types have just maintained the ancestral structures, which however does not provide sufficient evidence for their close relatedness. Regardless of whichever diverged first, MIKC*-, MIKC<sup>C</sup>- or M-type, all scenarios equally assume an ancestral gain of the K domain and a subsequent specific loss in the M-type lineage (Fig. 1a).</p><p>To reappraise the origin of MIKC*-type TFs and to resolve the relatedness among MIKC*-, MIKC<sup>C</sup>- and M-types, we performed phylogenetic analyses with broad sampling coverage across major lineages of land plants (Supporting Information Table S1). We aligned the MADS domain amino acid sequences of all MADS-box TFs from six bryophytes, four lycophytes, four ferns, five gymnosperms and three angiosperms (Figs 1b, S1; Table S2), with sequences from charophytes, the closest successive sister lineages of land plants, and green algae as outgroups. In the maximum-likelihood tree, the MIKC*-type TFs form a well-supported clade with Type I (M-type) TFs. We additionally generated a maximum-likelihood tree based on the codon alignments, which largely supports the new topology (Fig. S2). Since the MADS-box gene family has undergone several rounds of gene duplications in plants (Nam <i>et al</i>., <span>2004</span>), there are many redundant sequences in the above analysis, and especially the MIKC<sup>C</sup>-type sequences are dominating the dataset. We therefore selected representative sequences of comparable sample sizes for the MIKC*-type, MIKC<sup>C</sup>-type and Type I clades from divergent TF subfamilies in all those species. Phylogenetic trees generated with both maximum-likelihood and Bayesian inference show that MIKC*-type TFs constitute the nearest sister clade to the Type I (M-type) TFs, instead of MIKC<sup>C</sup>-type (Figs S3, S4). These new phylogenetic analyses all suggest that in a recent streptophytic ancestor of land plants, an MIKC-type gene duplicated into the precursor of the MIKC<sup>C</sup> clade and a precursor for the MIKC*/M-type clade. This latter gene subsequently duplicated to give rise to the precursor of the MIKC* clade and the precursor of the M-type clade, which arose after the loss of the K-domain exons.</p><p>We noticed that not all the charophytic MADS-box sequences were positioned as successive sisters to all MADS-box sequences in land plants, but none of the charophytes we analyzed had a pair of MADS-box genes each clearly belonging to Type I or II clades. We thus refrain from concluding whether the charophytic lineages shared the abovementioned one or two rounds of MADS-box gene duplication. If the duplication took place in the early lineage of streptophytes, multiple independent gene losses must have occurred in the paraphyletic charophytes. We focused on the land plant MADS-box TFs and generated maximum-likelihood trees without sequences from charophytes (Fig. S5). With the sequences from chlorophytes as the outgroup, in land plants the MIKC* clade is still closer to the M-type clade rather than the MIKC<sup>C</sup> clade.</p><p>We carried out approximate unbiased (AU) tests (Shimodaira, <span>2002</span>) to compare the phylogenetic trees corresponding to competing hypotheses (Figs S6, S7). One topology represents our new phylogeny. The second one is a constraint phylogeny forcing the Type I clade to be the outgroup of the originally considered combined Type II clade of both MIKC<sup>C</sup>- and MIKC*-type, reflecting the previous MIKC monophyly model. We also created a third constraint phylogeny resembling an alternative MIKC paraphyly hypothesis, assuming MIKC*-type diverged before the MIKC<sup>C</sup>-type and Type I split. The AU tests suggest that the new phylogeny with monophyly of MIKC* and M-type is significantly better than the previous proposition of MIKC* and MIKC<sup>C</sup> monophyly. Although the assumed third topology for MIKC<sup>C</sup> and M-type monophyly is not completely rejected based on the topology tests, the new phylogeny is substantially better. Resolving the relationship between these clades was challenging, since the two inferred successive duplication events giving rise to the precursors of the three types likely took place in a very short time span in the history of streptophyte evolution, predating the split of bryophytes and vascular plants. This is reflected by low posterior probability supports in the Bayesian inferences (Fig. S4). Nevertheless, combining all available evidence from an up-to-date and comprehensive sampling and multiple analyses, we find the model that MIKC<sup>C</sup>-type TFs diverged first and the M-type arose by loss of K-domain after the later divergence from the MIKC*-type with the highest probability.</p><p>The new phylogeny with the revised MIKC* origin aids to reconstruct the model depicting the functional evolution of MADS-box genes in land plants (Fig. 2). As suggested by several charophytic MADS-box genes (Tanabe <i>et al</i>., <span>2005</span>), and the sole green algal MADS-box gene that has been functionally characterized, CsubMADS1 in the haploid <i>Coccomyxa subellipsoidea</i> (Nayar & Thangavel, <span>2021</span>), it is likely that ancestral MADS-box genes primarily served gametophytic functions, particularly in stress tolerance and gamete development. MIKC*-type genes likely have maintained the ancestral gametophytic function, consistent with expression and function of MIKC*-type genes in pollen, the male gametophyte (Verelst <i>et al</i>., <span>2007a</span>,<span>b</span>; Adamczyk & Fernandez, <span>2009</span>; Liu <i>et al</i>., <span>2013</span>). Interestingly, the single-copy MIKC* ortholog in the liverwort <i>Marchantia polymorpha</i> MpMADS1 can rescue mutants deficient of MIKC* in Arabidopsis (Zobell <i>et al</i>., <span>2010</span>), suggesting conservation of MIKC*-type function in male gametophytes across land plants. Supporting this hypothesis, MIKC*-type genes in mosses, lycophytes and ferns are preferentially associated with gametophytes, especially the structures bearing male gametophytes (Svensson <i>et al</i>., <span>2000</span>; Riese <i>et al</i>., <span>2005</span>; Zobell <i>et al</i>., <span>2010</span>; Kwantes <i>et al</i>., <span>2012</span>). In seed plants, MIKC*-type gene expression became nearly completely restricted to male gametophytes (Verelst <i>et al</i>., <span>2007a</span>,<span>b</span>; Adamczyk & Fernandez, <span>2009</span>; Liu <i>et al</i>., <span>2013</span>; Gramzow <i>et al</i>., <span>2014</span>; Gu <i>et al</i>., <span>2022</span>). Similarly, M-type MADS-box TFs are functionally important in female gametophytes in seed plants, as well as in the endosperm in flowering plants (Bemer <i>et al</i>., <span>2010</span>; Masiero <i>et al</i>., <span>2011</span>; Qiu & Köhler, <span>2022</span>), complementing the requirement of MADS-box function after MIKC*-type evolved to be male-specific. It has been proposed that the expansion of the MADS-box gene family contributed to the complexity of the plant body plan (Theissen <i>et al</i>., <span>1996</span>; Kaufmann <i>et al</i>., <span>2005</span>; Thangavel & Nayar, <span>2018</span>). This is most pronounced for the MIKC<sup>C</sup> clade, which is famous for its regulatory role in the patterning of reproductive organs (Smaczniak <i>et al</i>., <span>2012</span>; Theissen <i>et al</i>., <span>2016</span>). Nevertheless, the regulatory role of MIKC<sup>C</sup>-type TFs in sporophytes is assumed to be derived, since ancestral MADS-box genes likely did not have a sporophytic function. This is inferred by charophytic MADS-box genes, which are not expressed in the zygote, the only diploid phase of charophytes (Tanabe <i>et al</i>., <span>2005</span>). In comparison with the MIKC<sup>C</sup>-type subfamily, the copy number of MIKC*-type TFs remained moderately low (Kwantes <i>et al</i>., <span>2012</span>; Liu <i>et al</i>., <span>2013</span>). Likewise, the M-type TFs remained less duplicated and differentiated in vascular plants, until they evolved a function in the endosperm of angiosperms (Qiu & Köhler, <span>2022</span>). Thus, number and differentiation patterns of MIKC* and M-type MADS-box genes align with the simplicity of gametophytes in vascular plants. Conversely, in the gametophyte-dominant bryophytes, the MIKC*-type subfamily largely expanded in several moss species, and the M-type subfamily underwent lineage-specific expansion in the <i>Anthoceros</i> hornworts, which may have contributed to the considerably more complex structures of mosses and hornworts compared with liverworts (Zobell <i>et al</i>., <span>2010</span>; Table S2).</p><p>Together, our new results suggest a reclassification that moves MIKC*-type TFs into the Type I clade (Fig. 2). Based on this revised phylogeny, the updated Type I TFs are a clade of MADS-box genes that primarily preserved their ancestral function in gametophyte development. During the evolution of seed plants, MIKC*-type and M-type adopted male- and female-specific functions, respectively. In parallel, the refined Type II clade, comprised of typical MIKC<sup>C</sup>-type genes, gradually diverged to be the sporophytic MADS-box subfamily, which repeatedly duplicated and neofunctionalized to generate new genetic regulators underlying the diverse body architectures of sporophytes.</p><p>We extended the collection of genomes analyzed in Qiu <i>et al</i>. (<span>2023</span>) with more species that represent all major lineages of land plants for MADS-box protein identification (Table S1). Amino acid sequences of MADS-box proteins in Arabidopsis (retrieved from TAIR10, https://www.arabidopsis.org/) were used as queries in the <span>Blastp</span> program to search for MADS-box proteins in these additional genomes. The MADS domains from identified MADS-box proteins were extracted based on the alignments to the MADS domain entries in the Conserved Domain Database (Lu <i>et al</i>., <span>2020</span>) by the conserved domain search tool, CD-Search (Marchler-Bauer & Bryant, <span>2004</span>). We aligned the MADS domains with <span>Muscle</span> by the default settings (Edgar, <span>2004</span>). We also used the amino acid alignment as a guide to generate the corresponding codon alignments (Notes S1). We first included all identified MADS-box TFs from a series of representative land plants: Arabidopsis, rice and <i>Amborella trichopoda</i> (angiosperm); <i>Thuja plicata</i>, <i>Cycas panzhihuaensis</i>, <i>Ginkgo biloba</i>, <i>Gnetum luofuense</i> and <i>Welwitschia mirabilis</i> (gymnosperms); <i>Ceratopteris richardii</i>, <i>Salvinia cucullate</i>, <i>Adiantum capillus-veneris</i> and <i>Alsophila spinulosa</i> (ferns); <i>Selaginella moellendorffii</i>, <i>Isoetes taiwanensis</i>, <i>Diphasiastrum complanatum</i> and <i>Lycopodium clavatum</i> (lycophytes); <i>Physcomitrium patens</i>, <i>Ceratodon purpureus</i>, <i>Sphagnum fallax</i>, <i>Takakia lepidozioides</i>, <i>Marchantia polymorpha</i> and <i>Anthoceros angustus</i> (bryophytes) (Table S2). This enormous dataset contains many more MIKC<sup>C</sup>-type sequences compared with MIKC*- and M-types. Therefore, we generated a downsized dataset with selected sequences in the three major MADS-box TF subfamilies across land plants. For the purpose of elucidating the deep branching pattern between the three major MADS-box clades, we specifically chose the genes with high confidence to be classified into a certain type as the representative sequences based on previous knowledge and the large maximum-likelihood tree generated in this study.</p><p>We applied <span>Iq-Tree</span> 2 to generate maximum-likelihood trees (Minh <i>et al</i>., <span>2020</span>). The implemented <span>ModelFinder</span> (Kalyaanamoorthy <i>et al</i>., <span>2017</span>) determined the JTT substitution matrix (Jones <i>et al</i>., <span>1992</span>) to be the best substitution model in the tree inference for the large dataset of protein alignments and LG (Le & Gascuel, <span>2008</span>) for the reduced dataset. For the nucleotide alignments, we allowed different evolutionary rates between partitions of the first, second and third codon positions. We ran 1000 replicates of ultrafast bootstraps to estimate the support for reconstructed branches (Hoang <i>et al</i>., <span>2018</span>). We also employed <span>Phylobayes</span> (v.3.2) to perform Bayesian inference under the CAT+GTR model with two chains. After ensuring that the two chains had converged with a maxdiff < 0.3, a consensus tree was created. The effective sample sizes of the different parameters were verified to be greater than 200, except for α parameter of the gamma distribution of rates across sites, which was 78 (Lartillot <i>et al</i>., <span>2009</span>). We further compared the topology of constraint phylogenetic trees fitting the competing hypotheses, by topology tests such as AU tests (Shimodaira, <span>2002</span>) supported in <span>Iq-Tree</span> 2 (Minh <i>et al</i>., <span>2020</span>).</p><p>None declared.</p><p>YQ, ZL and CK planned and designed the research. YQ and ZL analyzed data. YQ, ZL and CK wrote the manuscript. YQ and ZL contributed equally to this work.</p>","PeriodicalId":214,"journal":{"name":"New Phytologist","volume":"244 2","pages":"358-363"},"PeriodicalIF":8.3000,"publicationDate":"2024-08-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/nph.20065","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"New Phytologist","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/nph.20065","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"PLANT SCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
MADS-box transcription factors (TFs) have gained widespread recognition for their exceptional diversity and pivotal roles in various biological functions across eukaryotic organisms. Specifically, in land plants, the MADS-box gene family has undergone substantial expansion and provided the genetic raw material for many developmental novelties, including flowers, fruits and seeds. Therefore, understanding the origin of MADS-box genes is crucial for gaining insights into the evolutionary success of land plants. Land plant MADS-box TFs have been categorized into two groups. MIKC-type (Type II) TFs are named after the structural arrangement of the MADS (M), Intervening (I) and plant-specific Keratin-like (K) domain, followed by a variable C-terminal region. By contrast, M-type (Type I) TFs lack the K domain (Alvarez-Buylla et al., 2000; Kofuji et al., 2003; Nam et al., 2004). The identification of mutants exhibiting distinct flower organ patterns led to the discovery of a multitude of MIKC-type MADS-box TFs in plants (Ng & Yanofsky, 2001; Nam et al., 2003; Kaufmann et al., 2005). By contrast, the identification of M-type genes occurred exclusively through bioinformatic analyses following the unveiling of the Arabidopsis thaliana genome (Alvarez-Buylla et al., 2000). Within the MIKC-type of MADS-box TFs, the MIKC*-type is distinguished based on the different arrangements of exons that encode the K domain (Svensson et al., 2000; Henschel et al., 2002; Zobell et al., 2010; Kwantes et al., 2012; Rümpler et al., 2023). Both the ‘classic’ MIKCC- and the MIKC*-types are present across land plant lineages and form well-supported separate clades (Kwantes et al., 2012; Gramzow & Theissen, 2013; Liu et al., 2013), suggesting that they diverged before the diversification of land plant lineages (Henschel et al., 2002; Kofuji et al., 2003; Tanabe et al., 2005; Kwantes et al., 2012).
It was a long-standing prevailing view in the field that plant Type II genes are orthologs to myocyte enhancer factor-2 (MEF2) genes in animals. Those were presumed to have diverged from plant Type I genes and animal serum response factor (SRF) genes via an ancient duplication before the divergence of the extant eukaryotic lineages (Alvarez-Buylla et al., 2000). Thus, with the shared presence of the K domain, MIKC*-type MADS-box genes were intuitively considered more closely related to MIKCC-type genes, collectively called Type II. This adheres to the principle of parsimony, by which the acquisition of the K domain should ideally have occurred only once during the evolution of plants (Kaufmann et al., 2005; Thangavel & Nayar, 2018) (Fig. 1a). However, we recently showed that both Type I and Type II genes are land plant-specific MEF2 orthologs that arose from a duplication event predating the origin of land plants (Qiu et al., 2023). Therefore, the plant Type I and Type II genes are more closely related than previously appreciated (Fig. 1a). Noticeably, the Type I and II gene duplication hypothetically took place around the time inferred for the rise of MIKC*-type genes (Kofuji et al., 2003; Kwantes et al., 2012). This raises the question whether the previously proposed monophyletic relationship between the MIKC*- and MIKCC-type as the Type II clade is better supported than the alternative hypothesis proposing that MIKC*-type genes are close paralogs of the Type I (M-type) clade (Fig. 1a). Since the ancestral land plant MADS-box gene giving rise to all current subfamilies has been inferred to include the K domain (Kaufmann et al., 2005; Thangavel & Nayar, 2018; Qiu et al., 2023), this domain is not only shared by MIKCC-type and MIKC*-type, but also inherited by the ancestral gene leading to the extant Type I clade. Therefore, MIKCC- and MIKC*-types have just maintained the ancestral structures, which however does not provide sufficient evidence for their close relatedness. Regardless of whichever diverged first, MIKC*-, MIKCC- or M-type, all scenarios equally assume an ancestral gain of the K domain and a subsequent specific loss in the M-type lineage (Fig. 1a).
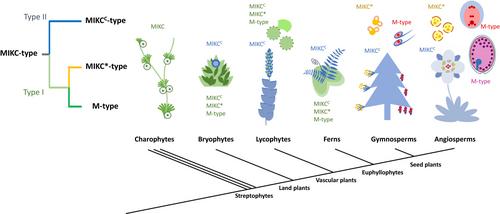
To reappraise the origin of MIKC*-type TFs and to resolve the relatedness among MIKC*-, MIKCC- and M-types, we performed phylogenetic analyses with broad sampling coverage across major lineages of land plants (Supporting Information Table S1). We aligned the MADS domain amino acid sequences of all MADS-box TFs from six bryophytes, four lycophytes, four ferns, five gymnosperms and three angiosperms (Figs 1b, S1; Table S2), with sequences from charophytes, the closest successive sister lineages of land plants, and green algae as outgroups. In the maximum-likelihood tree, the MIKC*-type TFs form a well-supported clade with Type I (M-type) TFs. We additionally generated a maximum-likelihood tree based on the codon alignments, which largely supports the new topology (Fig. S2). Since the MADS-box gene family has undergone several rounds of gene duplications in plants (Nam et al., 2004), there are many redundant sequences in the above analysis, and especially the MIKCC-type sequences are dominating the dataset. We therefore selected representative sequences of comparable sample sizes for the MIKC*-type, MIKCC-type and Type I clades from divergent TF subfamilies in all those species. Phylogenetic trees generated with both maximum-likelihood and Bayesian inference show that MIKC*-type TFs constitute the nearest sister clade to the Type I (M-type) TFs, instead of MIKCC-type (Figs S3, S4). These new phylogenetic analyses all suggest that in a recent streptophytic ancestor of land plants, an MIKC-type gene duplicated into the precursor of the MIKCC clade and a precursor for the MIKC*/M-type clade. This latter gene subsequently duplicated to give rise to the precursor of the MIKC* clade and the precursor of the M-type clade, which arose after the loss of the K-domain exons.
We noticed that not all the charophytic MADS-box sequences were positioned as successive sisters to all MADS-box sequences in land plants, but none of the charophytes we analyzed had a pair of MADS-box genes each clearly belonging to Type I or II clades. We thus refrain from concluding whether the charophytic lineages shared the abovementioned one or two rounds of MADS-box gene duplication. If the duplication took place in the early lineage of streptophytes, multiple independent gene losses must have occurred in the paraphyletic charophytes. We focused on the land plant MADS-box TFs and generated maximum-likelihood trees without sequences from charophytes (Fig. S5). With the sequences from chlorophytes as the outgroup, in land plants the MIKC* clade is still closer to the M-type clade rather than the MIKCC clade.
We carried out approximate unbiased (AU) tests (Shimodaira, 2002) to compare the phylogenetic trees corresponding to competing hypotheses (Figs S6, S7). One topology represents our new phylogeny. The second one is a constraint phylogeny forcing the Type I clade to be the outgroup of the originally considered combined Type II clade of both MIKCC- and MIKC*-type, reflecting the previous MIKC monophyly model. We also created a third constraint phylogeny resembling an alternative MIKC paraphyly hypothesis, assuming MIKC*-type diverged before the MIKCC-type and Type I split. The AU tests suggest that the new phylogeny with monophyly of MIKC* and M-type is significantly better than the previous proposition of MIKC* and MIKCC monophyly. Although the assumed third topology for MIKCC and M-type monophyly is not completely rejected based on the topology tests, the new phylogeny is substantially better. Resolving the relationship between these clades was challenging, since the two inferred successive duplication events giving rise to the precursors of the three types likely took place in a very short time span in the history of streptophyte evolution, predating the split of bryophytes and vascular plants. This is reflected by low posterior probability supports in the Bayesian inferences (Fig. S4). Nevertheless, combining all available evidence from an up-to-date and comprehensive sampling and multiple analyses, we find the model that MIKCC-type TFs diverged first and the M-type arose by loss of K-domain after the later divergence from the MIKC*-type with the highest probability.
The new phylogeny with the revised MIKC* origin aids to reconstruct the model depicting the functional evolution of MADS-box genes in land plants (Fig. 2). As suggested by several charophytic MADS-box genes (Tanabe et al., 2005), and the sole green algal MADS-box gene that has been functionally characterized, CsubMADS1 in the haploid Coccomyxa subellipsoidea (Nayar & Thangavel, 2021), it is likely that ancestral MADS-box genes primarily served gametophytic functions, particularly in stress tolerance and gamete development. MIKC*-type genes likely have maintained the ancestral gametophytic function, consistent with expression and function of MIKC*-type genes in pollen, the male gametophyte (Verelst et al., 2007a,b; Adamczyk & Fernandez, 2009; Liu et al., 2013). Interestingly, the single-copy MIKC* ortholog in the liverwort Marchantia polymorpha MpMADS1 can rescue mutants deficient of MIKC* in Arabidopsis (Zobell et al., 2010), suggesting conservation of MIKC*-type function in male gametophytes across land plants. Supporting this hypothesis, MIKC*-type genes in mosses, lycophytes and ferns are preferentially associated with gametophytes, especially the structures bearing male gametophytes (Svensson et al., 2000; Riese et al., 2005; Zobell et al., 2010; Kwantes et al., 2012). In seed plants, MIKC*-type gene expression became nearly completely restricted to male gametophytes (Verelst et al., 2007a,b; Adamczyk & Fernandez, 2009; Liu et al., 2013; Gramzow et al., 2014; Gu et al., 2022). Similarly, M-type MADS-box TFs are functionally important in female gametophytes in seed plants, as well as in the endosperm in flowering plants (Bemer et al., 2010; Masiero et al., 2011; Qiu & Köhler, 2022), complementing the requirement of MADS-box function after MIKC*-type evolved to be male-specific. It has been proposed that the expansion of the MADS-box gene family contributed to the complexity of the plant body plan (Theissen et al., 1996; Kaufmann et al., 2005; Thangavel & Nayar, 2018). This is most pronounced for the MIKCC clade, which is famous for its regulatory role in the patterning of reproductive organs (Smaczniak et al., 2012; Theissen et al., 2016). Nevertheless, the regulatory role of MIKCC-type TFs in sporophytes is assumed to be derived, since ancestral MADS-box genes likely did not have a sporophytic function. This is inferred by charophytic MADS-box genes, which are not expressed in the zygote, the only diploid phase of charophytes (Tanabe et al., 2005). In comparison with the MIKCC-type subfamily, the copy number of MIKC*-type TFs remained moderately low (Kwantes et al., 2012; Liu et al., 2013). Likewise, the M-type TFs remained less duplicated and differentiated in vascular plants, until they evolved a function in the endosperm of angiosperms (Qiu & Köhler, 2022). Thus, number and differentiation patterns of MIKC* and M-type MADS-box genes align with the simplicity of gametophytes in vascular plants. Conversely, in the gametophyte-dominant bryophytes, the MIKC*-type subfamily largely expanded in several moss species, and the M-type subfamily underwent lineage-specific expansion in the Anthoceros hornworts, which may have contributed to the considerably more complex structures of mosses and hornworts compared with liverworts (Zobell et al., 2010; Table S2).
Together, our new results suggest a reclassification that moves MIKC*-type TFs into the Type I clade (Fig. 2). Based on this revised phylogeny, the updated Type I TFs are a clade of MADS-box genes that primarily preserved their ancestral function in gametophyte development. During the evolution of seed plants, MIKC*-type and M-type adopted male- and female-specific functions, respectively. In parallel, the refined Type II clade, comprised of typical MIKCC-type genes, gradually diverged to be the sporophytic MADS-box subfamily, which repeatedly duplicated and neofunctionalized to generate new genetic regulators underlying the diverse body architectures of sporophytes.
We extended the collection of genomes analyzed in Qiu et al. (2023) with more species that represent all major lineages of land plants for MADS-box protein identification (Table S1). Amino acid sequences of MADS-box proteins in Arabidopsis (retrieved from TAIR10, https://www.arabidopsis.org/) were used as queries in the Blastp program to search for MADS-box proteins in these additional genomes. The MADS domains from identified MADS-box proteins were extracted based on the alignments to the MADS domain entries in the Conserved Domain Database (Lu et al., 2020) by the conserved domain search tool, CD-Search (Marchler-Bauer & Bryant, 2004). We aligned the MADS domains with Muscle by the default settings (Edgar, 2004). We also used the amino acid alignment as a guide to generate the corresponding codon alignments (Notes S1). We first included all identified MADS-box TFs from a series of representative land plants: Arabidopsis, rice and Amborella trichopoda (angiosperm); Thuja plicata, Cycas panzhihuaensis, Ginkgo biloba, Gnetum luofuense and Welwitschia mirabilis (gymnosperms); Ceratopteris richardii, Salvinia cucullate, Adiantum capillus-veneris and Alsophila spinulosa (ferns); Selaginella moellendorffii, Isoetes taiwanensis, Diphasiastrum complanatum and Lycopodium clavatum (lycophytes); Physcomitrium patens, Ceratodon purpureus, Sphagnum fallax, Takakia lepidozioides, Marchantia polymorpha and Anthoceros angustus (bryophytes) (Table S2). This enormous dataset contains many more MIKCC-type sequences compared with MIKC*- and M-types. Therefore, we generated a downsized dataset with selected sequences in the three major MADS-box TF subfamilies across land plants. For the purpose of elucidating the deep branching pattern between the three major MADS-box clades, we specifically chose the genes with high confidence to be classified into a certain type as the representative sequences based on previous knowledge and the large maximum-likelihood tree generated in this study.
We applied Iq-Tree 2 to generate maximum-likelihood trees (Minh et al., 2020). The implemented ModelFinder (Kalyaanamoorthy et al., 2017) determined the JTT substitution matrix (Jones et al., 1992) to be the best substitution model in the tree inference for the large dataset of protein alignments and LG (Le & Gascuel, 2008) for the reduced dataset. For the nucleotide alignments, we allowed different evolutionary rates between partitions of the first, second and third codon positions. We ran 1000 replicates of ultrafast bootstraps to estimate the support for reconstructed branches (Hoang et al., 2018). We also employed Phylobayes (v.3.2) to perform Bayesian inference under the CAT+GTR model with two chains. After ensuring that the two chains had converged with a maxdiff < 0.3, a consensus tree was created. The effective sample sizes of the different parameters were verified to be greater than 200, except for α parameter of the gamma distribution of rates across sites, which was 78 (Lartillot et al., 2009). We further compared the topology of constraint phylogenetic trees fitting the competing hypotheses, by topology tests such as AU tests (Shimodaira, 2002) supported in Iq-Tree 2 (Minh et al., 2020).
None declared.
YQ, ZL and CK planned and designed the research. YQ and ZL analyzed data. YQ, ZL and CK wrote the manuscript. YQ and ZL contributed equally to this work.
期刊介绍:
New Phytologist is an international electronic journal published 24 times a year. It is owned by the New Phytologist Foundation, a non-profit-making charitable organization dedicated to promoting plant science. The journal publishes excellent, novel, rigorous, and timely research and scholarship in plant science and its applications. The articles cover topics in five sections: Physiology & Development, Environment, Interaction, Evolution, and Transformative Plant Biotechnology. These sections encompass intracellular processes, global environmental change, and encourage cross-disciplinary approaches. The journal recognizes the use of techniques from molecular and cell biology, functional genomics, modeling, and system-based approaches in plant science. Abstracting and Indexing Information for New Phytologist includes Academic Search, AgBiotech News & Information, Agroforestry Abstracts, Biochemistry & Biophysics Citation Index, Botanical Pesticides, CAB Abstracts®, Environment Index, Global Health, and Plant Breeding Abstracts, and others.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
