José Manuel Guevara-Vela, Miguel Gallegos, Tomás Rocha-Rinza, Álvaro Muñoz-Castro, Peter L. Rodríguez Kessler, Ángel Martín Pendás
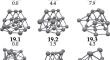
{"title":"New global minimum conformers for the Pt\\(_{19}\\) and Pt\\(_{20}\\) clusters: low symmetric species featuring different active sites","authors":"José Manuel Guevara-Vela, Miguel Gallegos, Tomás Rocha-Rinza, Álvaro Muñoz-Castro, Peter L. Rodríguez Kessler, Ángel Martín Pendás","doi":"10.1007/s00894-024-06099-5","DOIUrl":null,"url":null,"abstract":"<div><h3>\n <b>Context</b>\n </h3><p>The study of platinum (Pt) clusters and nanoparticles is essential due to their extensive range of potential technological applications, particularly in catalysis. The electronic properties that yield optimal catalytic performance at the nanoscale are significantly influenced by the size and structure of Pt clusters. This research aimed to identify the lowest-energy conformers for Pt<span>\\(_{18}\\)</span>, Pt<span>\\(_{19}\\)</span>, and Pt<span>\\(_{20}\\)</span> species using Density Functional Theory (DFT). We discovered new low-symmetry conformers for Pt<span>\\(_{19}\\)</span> and Pt<span>\\(_{20}\\)</span>, which are 3.0 and 1.0 kcal/mol more stable, respectively, than previously reported structures. Our study highlights the importance of using density functional approximations that incorporate moderate levels of exact Hartree-Fock exchange, alongside basis sets of at least quadruple-zeta quality. The resulting structures are asymmetric with varying active sites, as evidenced by sigma hole analysis on the electrostatic potential surface. This suggests a potential correlation between electronic structure and catalytic properties, warranting further investigation.</p><h3>\n <b>Methods</b>\n </h3><p>An equivariant graph neural network interatomic potential (NequIP) within the Atomic Simulation Environment suite (ASE) was used to provide initial geometries of the aggregates under study. DFT calculations were performed with the ORCA 5 package, using functional approximations that included Generalized Gradient Approximation (PBE), meta-GGA (TPSS, M06-L), hybrid (PBE0, PBEh), meta-GGA hybrid (TPSSh), and range-separated hybrid (<span>\\(\\omega \\)</span>B97x) functionals. Def2-TZVP and Def2-QZVP as well as members of the cc-pwCVXZ-PP family to check basis set convergence were used. QTAIM calculations were performed using the AIMAll suite. Structures were visualized with the AVOGADRO code.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"30 9","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2024-08-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11330413/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-024-06099-5","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Context
The study of platinum (Pt) clusters and nanoparticles is essential due to their extensive range of potential technological applications, particularly in catalysis. The electronic properties that yield optimal catalytic performance at the nanoscale are significantly influenced by the size and structure of Pt clusters. This research aimed to identify the lowest-energy conformers for Pt\(_{18}\), Pt\(_{19}\), and Pt\(_{20}\) species using Density Functional Theory (DFT). We discovered new low-symmetry conformers for Pt\(_{19}\) and Pt\(_{20}\), which are 3.0 and 1.0 kcal/mol more stable, respectively, than previously reported structures. Our study highlights the importance of using density functional approximations that incorporate moderate levels of exact Hartree-Fock exchange, alongside basis sets of at least quadruple-zeta quality. The resulting structures are asymmetric with varying active sites, as evidenced by sigma hole analysis on the electrostatic potential surface. This suggests a potential correlation between electronic structure and catalytic properties, warranting further investigation.
Methods
An equivariant graph neural network interatomic potential (NequIP) within the Atomic Simulation Environment suite (ASE) was used to provide initial geometries of the aggregates under study. DFT calculations were performed with the ORCA 5 package, using functional approximations that included Generalized Gradient Approximation (PBE), meta-GGA (TPSS, M06-L), hybrid (PBE0, PBEh), meta-GGA hybrid (TPSSh), and range-separated hybrid (\(\omega \)B97x) functionals. Def2-TZVP and Def2-QZVP as well as members of the cc-pwCVXZ-PP family to check basis set convergence were used. QTAIM calculations were performed using the AIMAll suite. Structures were visualized with the AVOGADRO code.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
