Chelsea D. Villanueva, Markéta Bohunická, Jeffrey R. Johansen
{"title":"We are doing it wrong: Putting homology before phylogeny in cyanobacterial taxonomy","authors":"Chelsea D. Villanueva, Markéta Bohunická, Jeffrey R. Johansen","doi":"10.1111/jpy.13491","DOIUrl":null,"url":null,"abstract":"<p>The rapid expansion of whole genome sequencing in bacterial taxonomy has revealed deep evolutionary relationships and speciation signals, but assembly methods often miss true nucleotide diversity in the ribosomal operons. Though it lacks sufficient phylogenetic signal at the species level, the 16S ribosomal RNA gene is still much used in bacterial taxonomy. In cyanobacterial taxonomy, comparisons of 16S–23S Internal Transcribed Spacer (ITS) regions are used to bridge this information gap. Although ITS rRNA region analyses are routinely being used to identify species, researchers often do not identify orthologous operons, which leads to improper comparisons. No method for delineating orthologous operon copies from paralogous ones has been established. A new method for recognizing orthologous ribosomal operons by quantifying the conserved paired nucleotides in a helical domain of the ITS, has been developed. The D1′ Index quantifies differences in the ratio of pyrimidines to purines in paired nucleotide sequences of this helix. Comparing 111 operon sequences from 89 strains of <i>Brasilonema</i>, four orthologous operon types were identified. Plotting D1′ Index values against the length of helices produced clear separation of orthologs. Most orthologous operons in this study were observed both with and without tRNA genes present. We hypothesize that genomic rearrangement, not gene duplication, is responsible for the variation among orthologs. This new method will allow cyanobacterial taxonomists to utilize ITS rRNA region data more correctly, preventing erroneous taxonomic hypotheses. Moreover, this work could assist genomicists in identifying and preserving evident sequence variability in ribosomal operons, which is an important proxy for evolution in prokaryotes.</p>","PeriodicalId":16831,"journal":{"name":"Journal of Phycology","volume":"60 5","pages":"1071-1089"},"PeriodicalIF":3.4000,"publicationDate":"2024-08-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/jpy.13491","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Phycology","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/jpy.13491","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MARINE & FRESHWATER BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
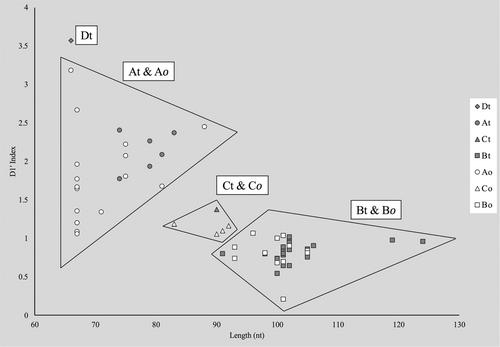
The rapid expansion of whole genome sequencing in bacterial taxonomy has revealed deep evolutionary relationships and speciation signals, but assembly methods often miss true nucleotide diversity in the ribosomal operons. Though it lacks sufficient phylogenetic signal at the species level, the 16S ribosomal RNA gene is still much used in bacterial taxonomy. In cyanobacterial taxonomy, comparisons of 16S–23S Internal Transcribed Spacer (ITS) regions are used to bridge this information gap. Although ITS rRNA region analyses are routinely being used to identify species, researchers often do not identify orthologous operons, which leads to improper comparisons. No method for delineating orthologous operon copies from paralogous ones has been established. A new method for recognizing orthologous ribosomal operons by quantifying the conserved paired nucleotides in a helical domain of the ITS, has been developed. The D1′ Index quantifies differences in the ratio of pyrimidines to purines in paired nucleotide sequences of this helix. Comparing 111 operon sequences from 89 strains of Brasilonema, four orthologous operon types were identified. Plotting D1′ Index values against the length of helices produced clear separation of orthologs. Most orthologous operons in this study were observed both with and without tRNA genes present. We hypothesize that genomic rearrangement, not gene duplication, is responsible for the variation among orthologs. This new method will allow cyanobacterial taxonomists to utilize ITS rRNA region data more correctly, preventing erroneous taxonomic hypotheses. Moreover, this work could assist genomicists in identifying and preserving evident sequence variability in ribosomal operons, which is an important proxy for evolution in prokaryotes.
期刊介绍:
The Journal of Phycology was founded in 1965 by the Phycological Society of America. All aspects of basic and applied research on algae are included to provide a common medium for the ecologist, physiologist, cell biologist, molecular biologist, morphologist, oceanographer, taxonomist, geneticist, and biochemist. The Journal also welcomes research that emphasizes algal interactions with other organisms and the roles of algae as components of natural ecosystems.
All aspects of basic and applied research on algae are included to provide a common medium for the ecologist, physiologist, cell biologist, molecular biologist, morphologist, oceanographer, acquaculturist, systematist, geneticist, and biochemist. The Journal also welcomes research that emphasizes algal interactions with other organisms and the roles of algae as components of natural ecosystems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
