Jay S. McDonald-Ramos , Ian K. Hicklin , Zhaomin Yang , Anne M. Brown
{"title":"Identification of small molecule inhibitors of the Chloracidobacterium thermophilum type IV pilus protein PilB by ensemble virtual screening","authors":"Jay S. McDonald-Ramos , Ian K. Hicklin , Zhaomin Yang , Anne M. Brown","doi":"10.1016/j.abb.2024.110127","DOIUrl":null,"url":null,"abstract":"<div><p>Antivirulence strategy has been explored as an alternative to traditional antibiotic development. The bacterial type IV pilus is a virulence factor involved in host invasion and colonization in many antibiotic resistant pathogens. The PilB ATPase hydrolyzes ATP to drive the assembly of the pilus filament from pilin subunits. We evaluated <em>Chloracidobacterium thermophilum</em> PilB (<em>Ct</em>PilB) as a model for structure-based virtual screening by molecular docking and molecular dynamics (MD) simulations. A hexameric structure of <em>Ct</em>PilB was generated through homology modeling based on an existing crystal structure of a PilB from <em>Geobacter metallireducens</em>. Four representative structures were obtained from molecular dynamics simulations to examine the conformational plasticity of PilB and improve docking analyses by ensemble docking. Structural analyses after 1 μs of simulation revealed conformational changes in individual PilB subunits are dependent on ligand presence. Further, ensemble virtual screening of a library of 4234 compounds retrieved from the ZINC15 database identified five promising PilB inhibitors. Molecular docking and binding analyses using the four representative structures from MD simulations revealed that top-ranked compounds interact with multiple Walker A residues, one Asp-box residue, and one arginine finger, indicating these are key residues in inhibitor binding within the ATP binding pocket. The use of multiple conformations in molecular screening can provide greater insight into compound flexibility within receptor sites and better inform future drug development for therapeutics targeting the type IV pilus assembly ATPase.</p></div>","PeriodicalId":8174,"journal":{"name":"Archives of biochemistry and biophysics","volume":"760 ","pages":"Article 110127"},"PeriodicalIF":3.0000,"publicationDate":"2024-08-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S0003986124002492/pdfft?md5=8129562d1f8578e6c794166cd6c848c7&pid=1-s2.0-S0003986124002492-main.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Archives of biochemistry and biophysics","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0003986124002492","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
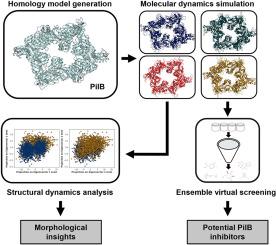
Antivirulence strategy has been explored as an alternative to traditional antibiotic development. The bacterial type IV pilus is a virulence factor involved in host invasion and colonization in many antibiotic resistant pathogens. The PilB ATPase hydrolyzes ATP to drive the assembly of the pilus filament from pilin subunits. We evaluated Chloracidobacterium thermophilum PilB (CtPilB) as a model for structure-based virtual screening by molecular docking and molecular dynamics (MD) simulations. A hexameric structure of CtPilB was generated through homology modeling based on an existing crystal structure of a PilB from Geobacter metallireducens. Four representative structures were obtained from molecular dynamics simulations to examine the conformational plasticity of PilB and improve docking analyses by ensemble docking. Structural analyses after 1 μs of simulation revealed conformational changes in individual PilB subunits are dependent on ligand presence. Further, ensemble virtual screening of a library of 4234 compounds retrieved from the ZINC15 database identified five promising PilB inhibitors. Molecular docking and binding analyses using the four representative structures from MD simulations revealed that top-ranked compounds interact with multiple Walker A residues, one Asp-box residue, and one arginine finger, indicating these are key residues in inhibitor binding within the ATP binding pocket. The use of multiple conformations in molecular screening can provide greater insight into compound flexibility within receptor sites and better inform future drug development for therapeutics targeting the type IV pilus assembly ATPase.
期刊介绍:
Archives of Biochemistry and Biophysics publishes quality original articles and reviews in the developing areas of biochemistry and biophysics.
Research Areas Include:
• Enzyme and protein structure, function, regulation. Folding, turnover, and post-translational processing
• Biological oxidations, free radical reactions, redox signaling, oxygenases, P450 reactions
• Signal transduction, receptors, membrane transport, intracellular signals. Cellular and integrated metabolism.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
