André Dallmann, Peter L. Bonate, Janelle Burnham, Blessy George, Lynne Yao, Jane Knöchel
{"title":"Enhancing inclusivity in clinical trials: Model-informed drug development for pregnant individuals in the era of personalized medicine","authors":"André Dallmann, Peter L. Bonate, Janelle Burnham, Blessy George, Lynne Yao, Jane Knöchel","doi":"10.1002/psp4.13218","DOIUrl":null,"url":null,"abstract":"<p>For decades, the administration of medication to pregnant and lactating individuals has occurred and the majority of pregnant individuals commonly receive medication during pregnancy. However, the inclusion of pregnant individuals is limited or is significantly underrepresented in global clinical trial research. Factors that may contribute to this gap include hesitancy of healthcare providers and patients, complex trial designs, ethical concerns, and the potential risk to the pregnant individual and fetus.<span><sup>1</sup></span> Consequently, pregnant and lactating individuals are prescribed potentially beneficial medicines with limited safety and efficacy information or guidance on optimal dosing for this patient population. Thus, it is vital to include pregnant individuals in the drug development process and engage early with global regulatory agencies.</p><p>Model-informed drug development (MIDD) methods are a selection of various quantitative methods that help to balance the risks and benefits of drug products in development. As such, these techniques are paramount to maximize the number of safe and effective medicines for pregnant individuals. Here, we discuss a roadmap of how each MIDD method (Figure 1) can be used to address the various challenges faced in this vulnerable patient population.</p><p>As stated earlier, pregnant individuals have historically been excluded from clinical therapeutics development trials and continue to be underrepresented in research. Importantly, failure to establish the correct dose/dosing regimen and the safety of treatments used during pregnancy may compromise the health of pregnant individuals and their fetuses. Under certain circumstances, it is ethically justifiable to include pregnant individuals in clinical trials in both the premarketing and postmarketing setting.<span><sup>7</sup></span> Additionally, it may also be ethically justifiable to obtain information on individuals who become pregnant while enrolled in a clinical trial. For example, if an individual becomes pregnant while on an investigational agent, they may consent to the collection of pharmacokinetic data that can be used to identify any changes in dosing that may be needed during pregnancy. However, at the time of marketing approval, there is generally little to no human data to inform the safety of drugs and biological products when used during pregnancy. Consequently, the FDA has the authority to issue postmarketing required (PMR) studies to collect information on the safety of medicines used during pregnancy. PMR studies are considered during the review of a marketing application and may be issued for treatments that will be used in females of reproductive potential when there is a need for data to inform on the safety of the use of the treatment during pregnancy.<span><sup>8</sup></span> In a recent review, only 16% of drugs that may be used in females of reproductive potential were issued PMRs for pregnancy (and/or lactation) studies.<span><sup>9</sup></span> However, there has been increasing stakeholder interest in the importance of collecting data in pregnant individuals as illustrated by the recommendations of the Task Force on Research Specific to Pregnant Women and Lactating Women (PRGLAC). The PRGLAC report included 15 recommendations to increase the availability of safe and effective therapies specific to pregnant and lactating individuals.<span><sup>10</sup></span></p><p>Although diversity and inclusion in clinical trials and personalized medicine are current paradigms, there is a lack of information about drug safety and efficacy in pregnant individuals. Various stakeholder efforts, further outlined in Table 1, strive to bridge this gap. While incentives for conducting clinical trials in pregnant individuals remain limited, regulatory agencies are increasingly emphasizing the need for PMR studies to gather essential information. To advance drug development in this population, it is crucial to engage early with regulatory agencies and explore MIDD tools which present promising avenues to broaden the access of pregnant individuals to the benefits of clinical research.</p><p>No funding was received for this work.</p><p>J.K. is an employee of AstraZeneca and owns stock. All other authors declared no competing interests for this work.</p><p>The views expressed in this perspective are the authors' views alone. They do not reflect the official views or guidance from the US Food and Drug Administration, nor should they be construed as such.</p><p>As Editor-in-Training of <i>CPT: Pharmacometrics & Systems Pharmacology</i> Jane Knöchel was not involved in the review or decision process for this paper.</p>","PeriodicalId":10774,"journal":{"name":"CPT: Pharmacometrics & Systems Pharmacology","volume":"13 11","pages":"1824-1829"},"PeriodicalIF":3.0000,"publicationDate":"2024-08-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/psp4.13218","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"CPT: Pharmacometrics & Systems Pharmacology","FirstCategoryId":"3","ListUrlMain":"https://ascpt.onlinelibrary.wiley.com/doi/10.1002/psp4.13218","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"PHARMACOLOGY & PHARMACY","Score":null,"Total":0}
引用次数: 0
Abstract
For decades, the administration of medication to pregnant and lactating individuals has occurred and the majority of pregnant individuals commonly receive medication during pregnancy. However, the inclusion of pregnant individuals is limited or is significantly underrepresented in global clinical trial research. Factors that may contribute to this gap include hesitancy of healthcare providers and patients, complex trial designs, ethical concerns, and the potential risk to the pregnant individual and fetus.1 Consequently, pregnant and lactating individuals are prescribed potentially beneficial medicines with limited safety and efficacy information or guidance on optimal dosing for this patient population. Thus, it is vital to include pregnant individuals in the drug development process and engage early with global regulatory agencies.
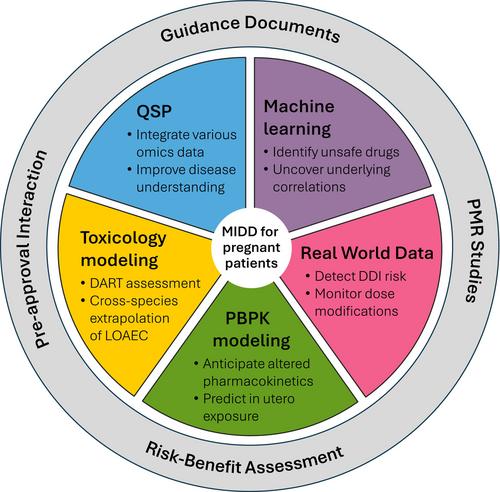
Model-informed drug development (MIDD) methods are a selection of various quantitative methods that help to balance the risks and benefits of drug products in development. As such, these techniques are paramount to maximize the number of safe and effective medicines for pregnant individuals. Here, we discuss a roadmap of how each MIDD method (Figure 1) can be used to address the various challenges faced in this vulnerable patient population.
As stated earlier, pregnant individuals have historically been excluded from clinical therapeutics development trials and continue to be underrepresented in research. Importantly, failure to establish the correct dose/dosing regimen and the safety of treatments used during pregnancy may compromise the health of pregnant individuals and their fetuses. Under certain circumstances, it is ethically justifiable to include pregnant individuals in clinical trials in both the premarketing and postmarketing setting.7 Additionally, it may also be ethically justifiable to obtain information on individuals who become pregnant while enrolled in a clinical trial. For example, if an individual becomes pregnant while on an investigational agent, they may consent to the collection of pharmacokinetic data that can be used to identify any changes in dosing that may be needed during pregnancy. However, at the time of marketing approval, there is generally little to no human data to inform the safety of drugs and biological products when used during pregnancy. Consequently, the FDA has the authority to issue postmarketing required (PMR) studies to collect information on the safety of medicines used during pregnancy. PMR studies are considered during the review of a marketing application and may be issued for treatments that will be used in females of reproductive potential when there is a need for data to inform on the safety of the use of the treatment during pregnancy.8 In a recent review, only 16% of drugs that may be used in females of reproductive potential were issued PMRs for pregnancy (and/or lactation) studies.9 However, there has been increasing stakeholder interest in the importance of collecting data in pregnant individuals as illustrated by the recommendations of the Task Force on Research Specific to Pregnant Women and Lactating Women (PRGLAC). The PRGLAC report included 15 recommendations to increase the availability of safe and effective therapies specific to pregnant and lactating individuals.10
Although diversity and inclusion in clinical trials and personalized medicine are current paradigms, there is a lack of information about drug safety and efficacy in pregnant individuals. Various stakeholder efforts, further outlined in Table 1, strive to bridge this gap. While incentives for conducting clinical trials in pregnant individuals remain limited, regulatory agencies are increasingly emphasizing the need for PMR studies to gather essential information. To advance drug development in this population, it is crucial to engage early with regulatory agencies and explore MIDD tools which present promising avenues to broaden the access of pregnant individuals to the benefits of clinical research.
No funding was received for this work.
J.K. is an employee of AstraZeneca and owns stock. All other authors declared no competing interests for this work.
The views expressed in this perspective are the authors' views alone. They do not reflect the official views or guidance from the US Food and Drug Administration, nor should they be construed as such.
As Editor-in-Training of CPT: Pharmacometrics & Systems Pharmacology Jane Knöchel was not involved in the review or decision process for this paper.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
