Dapeng Xiong, Kaicheng U, Jianfeng Sun, Adam P Cribbs
{"title":"PLMC: Language Model of Protein Sequences Enhances Protein Crystallization Prediction.","authors":"Dapeng Xiong, Kaicheng U, Jianfeng Sun, Adam P Cribbs","doi":"10.1007/s12539-024-00639-6","DOIUrl":null,"url":null,"abstract":"<p><p>X-ray diffraction crystallography has been most widely used for protein three-dimensional (3D) structure determination for which whether proteins are crystallizable is a central prerequisite. Yet, there are a number of procedures during protein crystallization, including protein material production, purification, and crystal production, which take turns affecting the crystallization outcome. Due to the expensive and laborious nature of this multi-stage process, various computational tools have been developed to predict protein crystallization propensity, which is then used to guide the experimental determination. In this study, we presented a novel deep learning framework, PLMC, to improve multi-stage protein crystallization propensity prediction by leveraging a pre-trained protein language model. To effectively train PLMC, two groups of features of each protein were integrated into a more comprehensive representation, including protein language embeddings from the large-scale protein sequence database and a handcrafted feature set consisting of physicochemical, sequence-based and disordered-related information. These features were further separately embedded for refinement, and then concatenated for the final prediction. Notably, our extensive benchmarking tests demonstrate that PLMC greatly outperforms other state-of-the-art methods by achieving AUC scores of 0.773, 0.893, and 0.913, respectively, at the aforementioned individual stages, and 0.982 at the final crystallization stage. Furthermore, PLMC is shown to be superior for predicting the crystallization of both globular and membrane proteins, as demonstrated by an AUC score of 0.991 for the latter. These results suggest the significant potential of PLMC in assisting researchers with the experimental design of crystallizable protein variants.</p>","PeriodicalId":13670,"journal":{"name":"Interdisciplinary Sciences: Computational Life Sciences","volume":" ","pages":"802-813"},"PeriodicalIF":3.9000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Interdisciplinary Sciences: Computational Life Sciences","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s12539-024-00639-6","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/19 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
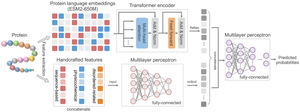
X-ray diffraction crystallography has been most widely used for protein three-dimensional (3D) structure determination for which whether proteins are crystallizable is a central prerequisite. Yet, there are a number of procedures during protein crystallization, including protein material production, purification, and crystal production, which take turns affecting the crystallization outcome. Due to the expensive and laborious nature of this multi-stage process, various computational tools have been developed to predict protein crystallization propensity, which is then used to guide the experimental determination. In this study, we presented a novel deep learning framework, PLMC, to improve multi-stage protein crystallization propensity prediction by leveraging a pre-trained protein language model. To effectively train PLMC, two groups of features of each protein were integrated into a more comprehensive representation, including protein language embeddings from the large-scale protein sequence database and a handcrafted feature set consisting of physicochemical, sequence-based and disordered-related information. These features were further separately embedded for refinement, and then concatenated for the final prediction. Notably, our extensive benchmarking tests demonstrate that PLMC greatly outperforms other state-of-the-art methods by achieving AUC scores of 0.773, 0.893, and 0.913, respectively, at the aforementioned individual stages, and 0.982 at the final crystallization stage. Furthermore, PLMC is shown to be superior for predicting the crystallization of both globular and membrane proteins, as demonstrated by an AUC score of 0.991 for the latter. These results suggest the significant potential of PLMC in assisting researchers with the experimental design of crystallizable protein variants.
期刊介绍:
Interdisciplinary Sciences--Computational Life Sciences aims to cover the most recent and outstanding developments in interdisciplinary areas of sciences, especially focusing on computational life sciences, an area that is enjoying rapid development at the forefront of scientific research and technology.
The journal publishes original papers of significant general interest covering recent research and developments. Articles will be published rapidly by taking full advantage of internet technology for online submission and peer-reviewing of manuscripts, and then by publishing OnlineFirstTM through SpringerLink even before the issue is built or sent to the printer.
The editorial board consists of many leading scientists with international reputation, among others, Luc Montagnier (UNESCO, France), Dennis Salahub (University of Calgary, Canada), Weitao Yang (Duke University, USA). Prof. Dongqing Wei at the Shanghai Jiatong University is appointed as the editor-in-chief; he made important contributions in bioinformatics and computational physics and is best known for his ground-breaking works on the theory of ferroelectric liquids. With the help from a team of associate editors and the editorial board, an international journal with sound reputation shall be created.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
