{"title":"Temperature effects on the branching dynamics in the model ambimodal (6 + 4)/(4 + 2) intramolecular cycloaddition reaction","authors":"Tatsuhiro Murakami, Daiki Hayashi, Yuya Kikuma, Keita Yamaki, Toshiyuki Takayanagi","doi":"10.1002/jcc.27484","DOIUrl":null,"url":null,"abstract":"<p>C<sub>14</sub>H<sub>20</sub> (tetradecapentaene) is a simple model system exhibiting post transition-state behavior, wherein both the (6 + 4) and (4 + 2) cycloaddition products are formed from one ambimocal transition state structure. We studied the bifurcation dynamics starting from the two ambimodal transition state structures, the chair-form and boat-form, using the quasi-classical trajectory, classical molecular dynamics, and ring-polymer molecular dynamics methods on the parameter-optimized semiempirical GFN2-xTB potential energy surface. It was found that the calculated branching fractions differ between the chair-form and boat-form due to the different nature in the IRC pathways. We also investigated the effects of temperature on bifurcation dynamics and found that, at higher temperatures, trajectories stay longer in the intermediate region of the potential energy surface.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 32","pages":"2778-2785"},"PeriodicalIF":4.8000,"publicationDate":"2024-08-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27484","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27484","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
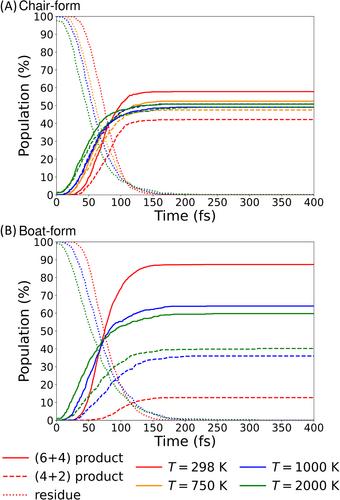
C14H20 (tetradecapentaene) is a simple model system exhibiting post transition-state behavior, wherein both the (6 + 4) and (4 + 2) cycloaddition products are formed from one ambimocal transition state structure. We studied the bifurcation dynamics starting from the two ambimodal transition state structures, the chair-form and boat-form, using the quasi-classical trajectory, classical molecular dynamics, and ring-polymer molecular dynamics methods on the parameter-optimized semiempirical GFN2-xTB potential energy surface. It was found that the calculated branching fractions differ between the chair-form and boat-form due to the different nature in the IRC pathways. We also investigated the effects of temperature on bifurcation dynamics and found that, at higher temperatures, trajectories stay longer in the intermediate region of the potential energy surface.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
