{"title":"Boosting the performance of molecular property prediction via graph–text alignment and multi-granularity representation enhancement","authors":"Zhuoran Zhao , Qing Zhou , Chengkai Wu , Renbin Su , Weihong Xiong","doi":"10.1016/j.jmgm.2024.108843","DOIUrl":null,"url":null,"abstract":"<div><p>Deep learning is playing an increasingly important role in accurate prediction of molecular properties. Prior to being processed by a deep learning model, a molecule is typically represented in the form of a text or a graph. While some methods attempt to integrate these two forms of molecular representations, the misalignment of graph and text embeddings presents a significant challenge to fuse two modalities. To solve this problem, we propose a method that aligns and fuses graph and text features in the embedding space by using contrastive loss and cross attentions. Additionally, we enhance the molecular representation by incorporating multi-granularity information of molecules on the levels of atoms, functional groups, and molecules. Extensive experiments show that our model outperforms state-of-the-art models in downstream tasks of molecular property prediction, achieving superior performance with less pretraining data. The source codes and data are available at <span><span>https://github.com/zzr624663649/multimodal_molecular_property</span><svg><path></path></svg></span>.</p></div>","PeriodicalId":16361,"journal":{"name":"Journal of molecular graphics & modelling","volume":"132 ","pages":"Article 108843"},"PeriodicalIF":3.0000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular graphics & modelling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1093326324001438","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/5 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
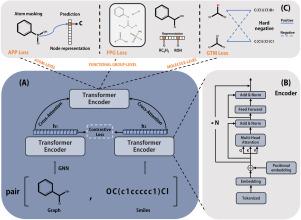
Deep learning is playing an increasingly important role in accurate prediction of molecular properties. Prior to being processed by a deep learning model, a molecule is typically represented in the form of a text or a graph. While some methods attempt to integrate these two forms of molecular representations, the misalignment of graph and text embeddings presents a significant challenge to fuse two modalities. To solve this problem, we propose a method that aligns and fuses graph and text features in the embedding space by using contrastive loss and cross attentions. Additionally, we enhance the molecular representation by incorporating multi-granularity information of molecules on the levels of atoms, functional groups, and molecules. Extensive experiments show that our model outperforms state-of-the-art models in downstream tasks of molecular property prediction, achieving superior performance with less pretraining data. The source codes and data are available at https://github.com/zzr624663649/multimodal_molecular_property.
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
