Jakub Kubečka, Daniel Ayoubi, Zeyuan Tang, Yosef Knattrup, Morten Engsvang, Haide Wu and Jonas Elm
{"title":"Accurate modeling of the potential energy surface of atmospheric molecular clusters boosted by neural networks†","authors":"Jakub Kubečka, Daniel Ayoubi, Zeyuan Tang, Yosef Knattrup, Morten Engsvang, Haide Wu and Jonas Elm","doi":"10.1039/D4VA00255E","DOIUrl":null,"url":null,"abstract":"<p >The computational cost of accurate quantum chemistry (QC) calculations of large molecular systems can often be unbearably high. Machine learning offers a lower computational cost compared to QC methods while maintaining their accuracy. In this study, we employ the polarizable atom interaction neural network (PaiNN) architecture to train and model the potential energy surface of molecular clusters relevant to atmospheric new particle formation, such as sulfuric acid–ammonia clusters. We compare the differences between PaiNN and previous kernel ridge regression modeling for the Clusteromics I–V data sets. We showcase three models capable of predicting electronic binding energies and interatomic forces with mean absolute errors of <0.3 kcal mol<small><sup>−1</sup></small> and <0.2 kcal mol<small><sup>−1</sup></small> Å<small><sup>−1</sup></small>, respectively. Furthermore, we demonstrate that the error of the modeled properties remains below the chemical accuracy of 1 kcal mol<small><sup>−1</sup></small> even for clusters vastly larger than those in the training database (up to (H<small><sub>2</sub></small>SO<small><sub>4</sub></small>)<small><sub>15</sub></small>(NH<small><sub>3</sub></small>)<small><sub>15</sub></small> clusters, containing 30 molecules). Consequently, we emphasize the potential applications of these models for faster and more thorough configurational sampling and for boosting molecular dynamics studies of large atmospheric molecular clusters.</p>","PeriodicalId":72941,"journal":{"name":"Environmental science. Advances","volume":" 10","pages":" 1438-1451"},"PeriodicalIF":4.4000,"publicationDate":"2024-08-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11334116/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Environmental science. Advances","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/va/d4va00255e","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"ENGINEERING, ENVIRONMENTAL","Score":null,"Total":0}
引用次数: 0
Abstract
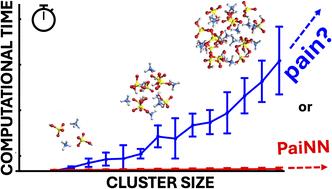
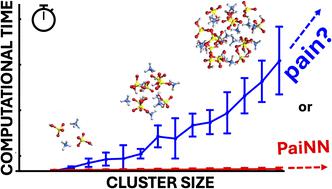
The computational cost of accurate quantum chemistry (QC) calculations of large molecular systems can often be unbearably high. Machine learning offers a lower computational cost compared to QC methods while maintaining their accuracy. In this study, we employ the polarizable atom interaction neural network (PaiNN) architecture to train and model the potential energy surface of molecular clusters relevant to atmospheric new particle formation, such as sulfuric acid–ammonia clusters. We compare the differences between PaiNN and previous kernel ridge regression modeling for the Clusteromics I–V data sets. We showcase three models capable of predicting electronic binding energies and interatomic forces with mean absolute errors of <0.3 kcal mol−1 and <0.2 kcal mol−1 Å−1, respectively. Furthermore, we demonstrate that the error of the modeled properties remains below the chemical accuracy of 1 kcal mol−1 even for clusters vastly larger than those in the training database (up to (H2SO4)15(NH3)15 clusters, containing 30 molecules). Consequently, we emphasize the potential applications of these models for faster and more thorough configurational sampling and for boosting molecular dynamics studies of large atmospheric molecular clusters.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
