{"title":"Data mining antibody sequences for database searching in bottom-up proteomics","authors":"Xuan-Tung Trinh , Rebecca Freitag , Konrad Krawczyk , Veit Schwämmle","doi":"10.1016/j.immuno.2024.100042","DOIUrl":null,"url":null,"abstract":"<div><p>Mass spectrometry-based proteomics facilitates the identification and quantification of thousands of proteins but encounters challenges in measuring human antibodies due to their vast diversity. Bottom-up proteomics methods primarily rely on database searches, comparing experimental peptide values to theoretical database sequences. While the human body can produce millions of distinct antibodies, current databases, such as UniProtKB/Swiss-Prot, contain only 1095 sequences (as of January 2024), potentially hindering antibody identification via mass spectrometry. Therefore, expanding the database is crucial for discovering new antibodies. Recent genomic studies have amassed millions of human antibody sequences in the Observed Antibody Space (OAS) database, yet this data remains underutilized. Leveraging this vast collection, we conduct efficient database searches in publicly available proteomics data, focusing on SARS-CoV-2. In our study, thirty million heavy antibody sequences from 146 SARS-CoV-2 patients in the OAS database were digested <em>in silico</em> to obtain 18 million unique peptides. These peptides form the basis for new bottom-up proteomics databases. We used those databases for searching new antibody peptides in publicly available SARS-CoV-2 human plasma samples in the Proteomics Identification Database (PRIDE). This approach avoids false positives in antibody peptide identification as confirmed by searching against negative controls (brain samples) and employing different database sizes. We show that new antibody peptides were found in previous plasma samples and expect that the newly discovered antibody peptides can be further employed to develop therapeutic antibodies. The method will be broadly applicable to find characteristic antibodies for other diseases.</p></div>","PeriodicalId":73343,"journal":{"name":"Immunoinformatics (Amsterdam, Netherlands)","volume":"15 ","pages":"Article 100042"},"PeriodicalIF":0.0000,"publicationDate":"2024-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S2667119024000120/pdfft?md5=6bc5ac01ada92397791db50d32ef768f&pid=1-s2.0-S2667119024000120-main.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Immunoinformatics (Amsterdam, Netherlands)","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2667119024000120","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/22 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
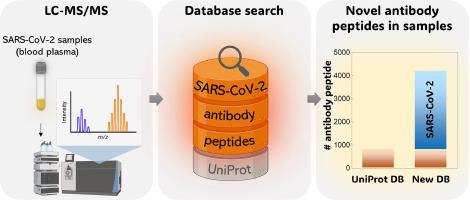
Abstract
Mass spectrometry-based proteomics facilitates the identification and quantification of thousands of proteins but encounters challenges in measuring human antibodies due to their vast diversity. Bottom-up proteomics methods primarily rely on database searches, comparing experimental peptide values to theoretical database sequences. While the human body can produce millions of distinct antibodies, current databases, such as UniProtKB/Swiss-Prot, contain only 1095 sequences (as of January 2024), potentially hindering antibody identification via mass spectrometry. Therefore, expanding the database is crucial for discovering new antibodies. Recent genomic studies have amassed millions of human antibody sequences in the Observed Antibody Space (OAS) database, yet this data remains underutilized. Leveraging this vast collection, we conduct efficient database searches in publicly available proteomics data, focusing on SARS-CoV-2. In our study, thirty million heavy antibody sequences from 146 SARS-CoV-2 patients in the OAS database were digested in silico to obtain 18 million unique peptides. These peptides form the basis for new bottom-up proteomics databases. We used those databases for searching new antibody peptides in publicly available SARS-CoV-2 human plasma samples in the Proteomics Identification Database (PRIDE). This approach avoids false positives in antibody peptide identification as confirmed by searching against negative controls (brain samples) and employing different database sizes. We show that new antibody peptides were found in previous plasma samples and expect that the newly discovered antibody peptides can be further employed to develop therapeutic antibodies. The method will be broadly applicable to find characteristic antibodies for other diseases.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
