Jae Hyeon Yu, Sangwon Lee, Yoon Jung Kim, Won Young Kim, Min Jung Lee, Yun Kim
{"title":"Assessing Post-Marketing Requirements for Orphan Drugs: A Cross-Sectional Analysis of FDA and EMA Oversight","authors":"Jae Hyeon Yu, Sangwon Lee, Yoon Jung Kim, Won Young Kim, Min Jung Lee, Yun Kim","doi":"10.1002/cpt.3397","DOIUrl":null,"url":null,"abstract":"<p>The U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) oversee pharmaceutical regulations, including orphan drugs targeting rare diseases with limited patient populations. Post-marketing studies are crucial for monitoring safety and efficacy, with post-marketing requirements (PMRs) mandated by the regulatory agencies to ensure compliance. This study aims to compare PMR statuses, objectives, and pivotal trial characteristics of orphan drugs approved by the FDA (<i>n</i> = 154) and EMA (<i>n</i> = 79) from 2008 to 2018, shedding light on regulatory differences and their impact on drug development. Contrary to expectations, our analysis found no significant disparity in the proportion of orphan drugs with and without PMRs approved by both the FDA (48.1%) and EMA (55.7%). Safety concerns surrounding orphan drugs post-approval, attributed partly to pivotal trial design, underscore the need for robust post-marketing surveillance. While the FDA primarily focuses on post-marketing safety (36.1%), the EMA places a higher emphasis on both efficacy and safety (47.1%), reflecting distinct approaches to PMR management between the two regulatory bodies. The observed trend of delayed PMRs at the EMA (47.1%) highlights the importance of effective cooperation between regulators and pharmaceutical companies to ensure the timely completion of PMRs and enhance drug safety.</p>","PeriodicalId":153,"journal":{"name":"Clinical Pharmacology & Therapeutics","volume":"116 6","pages":"1560-1571"},"PeriodicalIF":5.5000,"publicationDate":"2024-08-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/cpt.3397","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Pharmacology & Therapeutics","FirstCategoryId":"3","ListUrlMain":"https://ascpt.onlinelibrary.wiley.com/doi/10.1002/cpt.3397","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"PHARMACOLOGY & PHARMACY","Score":null,"Total":0}
引用次数: 0
Abstract
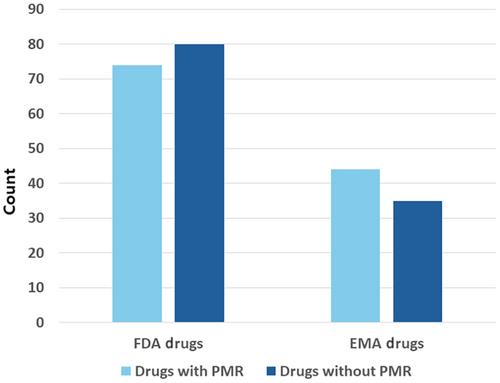
The U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) oversee pharmaceutical regulations, including orphan drugs targeting rare diseases with limited patient populations. Post-marketing studies are crucial for monitoring safety and efficacy, with post-marketing requirements (PMRs) mandated by the regulatory agencies to ensure compliance. This study aims to compare PMR statuses, objectives, and pivotal trial characteristics of orphan drugs approved by the FDA (n = 154) and EMA (n = 79) from 2008 to 2018, shedding light on regulatory differences and their impact on drug development. Contrary to expectations, our analysis found no significant disparity in the proportion of orphan drugs with and without PMRs approved by both the FDA (48.1%) and EMA (55.7%). Safety concerns surrounding orphan drugs post-approval, attributed partly to pivotal trial design, underscore the need for robust post-marketing surveillance. While the FDA primarily focuses on post-marketing safety (36.1%), the EMA places a higher emphasis on both efficacy and safety (47.1%), reflecting distinct approaches to PMR management between the two regulatory bodies. The observed trend of delayed PMRs at the EMA (47.1%) highlights the importance of effective cooperation between regulators and pharmaceutical companies to ensure the timely completion of PMRs and enhance drug safety.
期刊介绍:
Clinical Pharmacology & Therapeutics (CPT) is the authoritative cross-disciplinary journal in experimental and clinical medicine devoted to publishing advances in the nature, action, efficacy, and evaluation of therapeutics. CPT welcomes original Articles in the emerging areas of translational, predictive and personalized medicine; new therapeutic modalities including gene and cell therapies; pharmacogenomics, proteomics and metabolomics; bioinformation and applied systems biology complementing areas of pharmacokinetics and pharmacodynamics, human investigation and clinical trials, pharmacovigilence, pharmacoepidemiology, pharmacometrics, and population pharmacology.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
