Natsuki Watanabe, Yuta Hori, Hiroki Sugisawa, Tomonori Ida, Mitsuo Shoji, Yasuteru Shigeta
{"title":"A machine learning potential construction based on radial distribution function sampling","authors":"Natsuki Watanabe, Yuta Hori, Hiroki Sugisawa, Tomonori Ida, Mitsuo Shoji, Yasuteru Shigeta","doi":"10.1002/jcc.27497","DOIUrl":null,"url":null,"abstract":"<p>Sampling reference data is crucial in machine learning potential (MLP) construction. Inadequate coverage of local configurations in reference data may lead to unphysical behaviors in MLP-based molecular dynamics (MLP-MD) simulations. To address this problem, this study proposes a new on-the-fly reference data sampling method called radial distribution function (RDF)-based data sampling for MLP construction. This method detects and extracts anomalous structures from the trajectories of MLP-MD simulations by focusing on the shapes of RDFs. The detected structures are added to the reference data to improve the accuracy of the MLP. This method allows us to realize a reasonable MLP construction for liquid water with minimal additional data. We prepare data from an H<sub>2</sub>O molecular cluster system and verify whether the constructed MLPs are practical for bulk water systems. MLP-MD simulations without RDF-based data sampling show unphysical behaviors, such as atomic collisions. In contrast, after applying this method, we obtain MLP-MD trajectories with features, such as RDF shapes and angle distributions, that are comparable to those of ab initio MD simulations. Our simulation results demonstrate that the RDF-based data sampling approach is useful for constructing MLPs that are robust to extrapolations from molecular cluster systems to bulk systems without any specialized know-how.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 32","pages":"2949-2958"},"PeriodicalIF":4.8000,"publicationDate":"2024-09-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27497","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27497","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
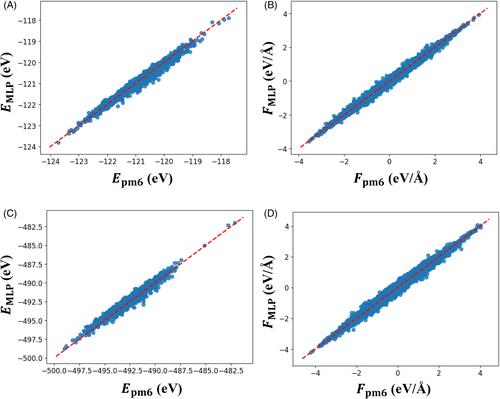
Sampling reference data is crucial in machine learning potential (MLP) construction. Inadequate coverage of local configurations in reference data may lead to unphysical behaviors in MLP-based molecular dynamics (MLP-MD) simulations. To address this problem, this study proposes a new on-the-fly reference data sampling method called radial distribution function (RDF)-based data sampling for MLP construction. This method detects and extracts anomalous structures from the trajectories of MLP-MD simulations by focusing on the shapes of RDFs. The detected structures are added to the reference data to improve the accuracy of the MLP. This method allows us to realize a reasonable MLP construction for liquid water with minimal additional data. We prepare data from an H2O molecular cluster system and verify whether the constructed MLPs are practical for bulk water systems. MLP-MD simulations without RDF-based data sampling show unphysical behaviors, such as atomic collisions. In contrast, after applying this method, we obtain MLP-MD trajectories with features, such as RDF shapes and angle distributions, that are comparable to those of ab initio MD simulations. Our simulation results demonstrate that the RDF-based data sampling approach is useful for constructing MLPs that are robust to extrapolations from molecular cluster systems to bulk systems without any specialized know-how.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
