Muhammad Saalim, Benjamin R. Clark, Peter R. Taylor
{"title":"Quantum chemical investigation of electronic transitions of mitorubrin azaphilones","authors":"Muhammad Saalim, Benjamin R. Clark, Peter R. Taylor","doi":"10.1002/jcc.27498","DOIUrl":null,"url":null,"abstract":"<p>Fungal azaphilones are a broad class of naturally-occurring pigments with diverse applications. Among the azaphilone pigments, mitorubrins are well recognized for their antiviral, antibacterial, antifungal, antiprotozoal, antidiabetic, and antiaging activities in addition to their well-known yellow-orange color. This makes these pigments interesting candidates for use in foods, as cosmetics, and as medicines. In particular, if it is desired to modify the properties of mitorubrin-based pigments, for example by derivatization, it is essential to have an understanding of the electronic spectra of the parent molecules. We have therefore undertaken a computational study of a series of mitorubrins, comparing our computed results with experimental UV/visible spectra. Both density-functional theory (DFT) and coupled-cluster (CC2) methods have been used, and in general, the results are in very good agreement with observation. In order to provide a simple and useful picture of the spectra we analyze the stronger transitions in terms of natural transition orbitals (NTOs).</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 32","pages":"2959-2968"},"PeriodicalIF":4.8000,"publicationDate":"2024-09-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27498","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
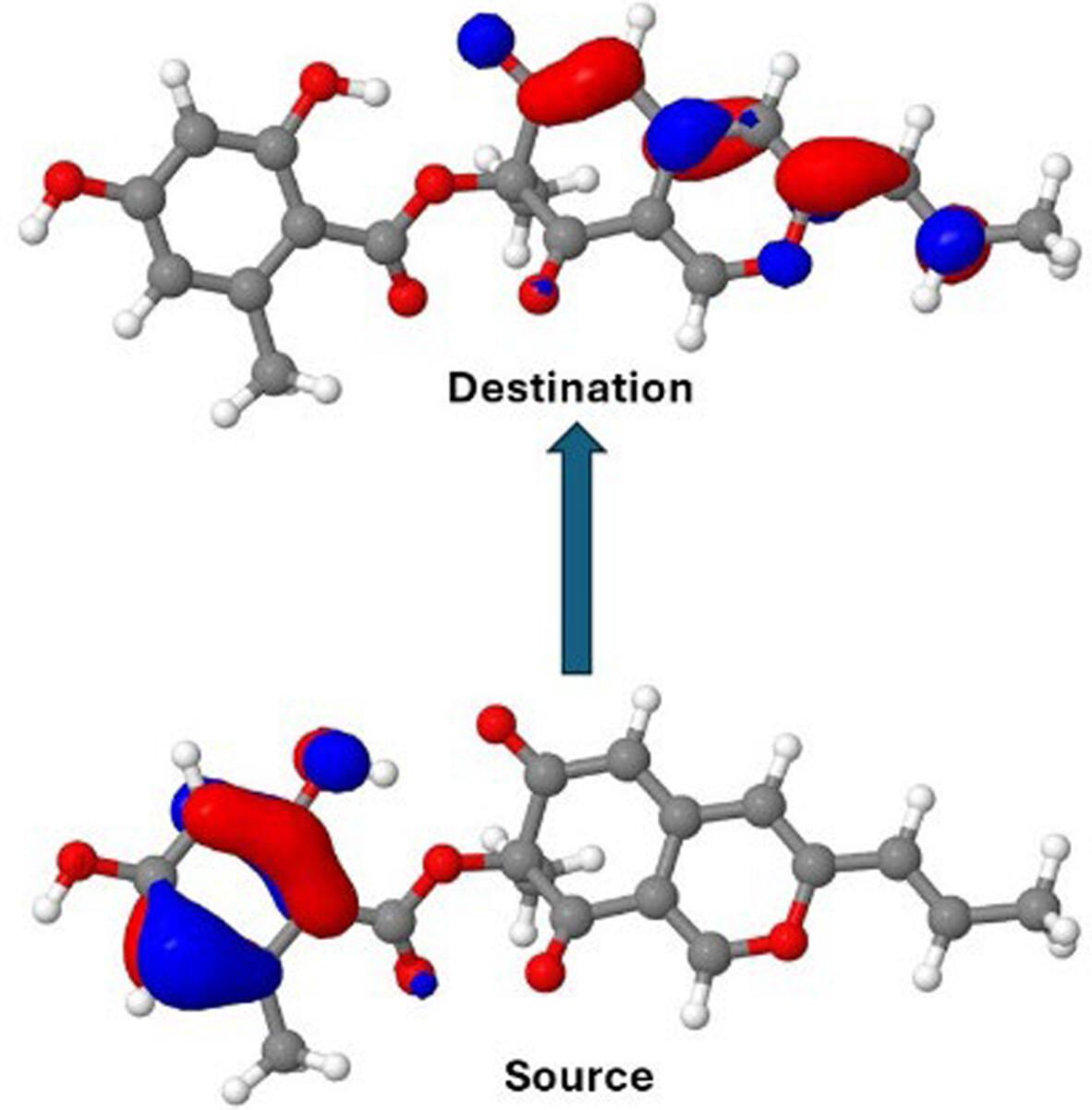
Fungal azaphilones are a broad class of naturally-occurring pigments with diverse applications. Among the azaphilone pigments, mitorubrins are well recognized for their antiviral, antibacterial, antifungal, antiprotozoal, antidiabetic, and antiaging activities in addition to their well-known yellow-orange color. This makes these pigments interesting candidates for use in foods, as cosmetics, and as medicines. In particular, if it is desired to modify the properties of mitorubrin-based pigments, for example by derivatization, it is essential to have an understanding of the electronic spectra of the parent molecules. We have therefore undertaken a computational study of a series of mitorubrins, comparing our computed results with experimental UV/visible spectra. Both density-functional theory (DFT) and coupled-cluster (CC2) methods have been used, and in general, the results are in very good agreement with observation. In order to provide a simple and useful picture of the spectra we analyze the stronger transitions in terms of natural transition orbitals (NTOs).
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
