{"title":"The origin of selectivity in the trimerization of 1,3-cyclopentadiene from an activation strain perspective","authors":"Ravshan S. Shamsiev, Nikolai A. Dontsenko","doi":"10.1007/s00894-024-06117-6","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>Quantum chemical modeling (DFT-PBE0/cc-pVTZ) of the [4 + 2]-cycloaddition reaction of 1,3-cyclopentadiene (CPD) to (exo/endo)-dicyclopentadiene (DCPD) was carried out, resulting in 14 products—CPD trimers. According to calculations, exo-addition of CPD to the norbornene (NB) fragment of DCPD and trans-addition of CPD to the cyclopentene (CP) fragment of DCPD are kinetically preferred. Ring strain energies <i>E</i><sub>RS</sub> were calculated for all trimers using the homodesmotic reaction approach. The least strained trimers are formed by exo-addition of CPD to the NB fragment of exo-DCPD, while the most strained ones are formed by endo-addition of CPD to the NB fragment of endo-DCPD. <i>E</i><sub>RS</sub> values are in good agreement with thermodynamic stability of trimers. Analysis of activation energy using the activation strain model showed steric effects causing deformation of the DCPD molecule upon reaching the transition state to be the leading factor of the magnitude of the cycloaddition reaction activation barrier. Deformation of the DCPD molecule mostly occurs in two dihedral angles—the angle of escape of H atoms from the plane of the double bond involved in cycloaddition and the angle between the NB and CP fragments. The sum of deviations of these angles in the transition states (or products) structures is in good agreement with Gibbs activation energies of cycloaddition reactions of CPD to DCPD.</p><h3>Methods</h3><p>Quantum chemical calculations were carried out using density functional theory in Gaussian 09 software. Hybrid exchange–correlation PBE0 functional was used with cc-pVTZ basis set.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"30 10","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2024-09-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-024-06117-6","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Context
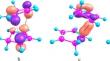
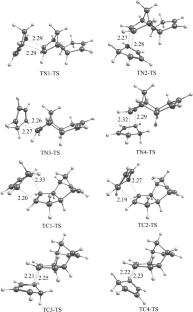
Quantum chemical modeling (DFT-PBE0/cc-pVTZ) of the [4 + 2]-cycloaddition reaction of 1,3-cyclopentadiene (CPD) to (exo/endo)-dicyclopentadiene (DCPD) was carried out, resulting in 14 products—CPD trimers. According to calculations, exo-addition of CPD to the norbornene (NB) fragment of DCPD and trans-addition of CPD to the cyclopentene (CP) fragment of DCPD are kinetically preferred. Ring strain energies ERS were calculated for all trimers using the homodesmotic reaction approach. The least strained trimers are formed by exo-addition of CPD to the NB fragment of exo-DCPD, while the most strained ones are formed by endo-addition of CPD to the NB fragment of endo-DCPD. ERS values are in good agreement with thermodynamic stability of trimers. Analysis of activation energy using the activation strain model showed steric effects causing deformation of the DCPD molecule upon reaching the transition state to be the leading factor of the magnitude of the cycloaddition reaction activation barrier. Deformation of the DCPD molecule mostly occurs in two dihedral angles—the angle of escape of H atoms from the plane of the double bond involved in cycloaddition and the angle between the NB and CP fragments. The sum of deviations of these angles in the transition states (or products) structures is in good agreement with Gibbs activation energies of cycloaddition reactions of CPD to DCPD.
Methods
Quantum chemical calculations were carried out using density functional theory in Gaussian 09 software. Hybrid exchange–correlation PBE0 functional was used with cc-pVTZ basis set.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
