{"title":"First principles study of perfluoroalkyl substance adsorption in M-MOF-74 metal organic frameworks","authors":"Daniel Mottern, Joshua Young","doi":"10.1016/j.commatsci.2024.113334","DOIUrl":null,"url":null,"abstract":"<div><p>Perfluoroalkyl substances (PFAS) are a family of chemical species consisting of a perfluorinated C-F bonded backbone, granting high thermal and aqueous stability. However, as they have been found to cause deleterious health effects in humans, their lack of degradation in air or water has led to the desire for new remediation technology, and absorptive removal by porous materials has been found to be a promising way to accomplish this. In this work, we investigate the metal organic framework (MOF) family known as <span><math><mi>M</mi></math></span>-MOF-74 (<span><math><mi>M</mi></math></span> = Cu, Mg, Zn, Pt) as potential adsorbents for the PFAS molecules PFOA, PFOS, and TFA. Using a combination of density functional theory (DFT) and <em>ab initio</em> molecular dynamics (AIMD) calculations, we find that protonated PFAS molecules can adsorb strongly in the <span><math><mi>M</mi></math></span>-MOF-74 frameworks, and that changing the <span><math><mi>M</mi></math></span> site results in tunability of the adsorption energy. Second, we find that, given the same length of the C backbone, those terminated by a -COOH group versus a -SO<span><math><msub><mrow></mrow><mrow><mn>3</mn></mrow></msub></math></span>H group binds more strongly; furthermore, the C backbone length has an effect as well, with long-chain PFAS adsorbing more strongly than short-chain. Finally, we find that deprotonated PFAS molecules do not interact with MOF compounds and display a positive adsorption energy, with Bader charge calculations show a distinct difference between protonated and deprotonated PFAS molecules. Through this work, we disentangle how MOF and PFAS chemistry affects adsorption in this family of compounds.</p></div>","PeriodicalId":10650,"journal":{"name":"Computational Materials Science","volume":"245 ","pages":"Article 113334"},"PeriodicalIF":3.3000,"publicationDate":"2024-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational Materials Science","FirstCategoryId":"88","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S092702562400555X","RegionNum":3,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/3 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"MATERIALS SCIENCE, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
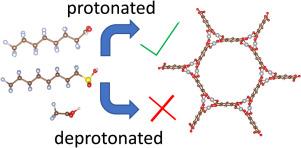
Perfluoroalkyl substances (PFAS) are a family of chemical species consisting of a perfluorinated C-F bonded backbone, granting high thermal and aqueous stability. However, as they have been found to cause deleterious health effects in humans, their lack of degradation in air or water has led to the desire for new remediation technology, and absorptive removal by porous materials has been found to be a promising way to accomplish this. In this work, we investigate the metal organic framework (MOF) family known as -MOF-74 ( = Cu, Mg, Zn, Pt) as potential adsorbents for the PFAS molecules PFOA, PFOS, and TFA. Using a combination of density functional theory (DFT) and ab initio molecular dynamics (AIMD) calculations, we find that protonated PFAS molecules can adsorb strongly in the -MOF-74 frameworks, and that changing the site results in tunability of the adsorption energy. Second, we find that, given the same length of the C backbone, those terminated by a -COOH group versus a -SOH group binds more strongly; furthermore, the C backbone length has an effect as well, with long-chain PFAS adsorbing more strongly than short-chain. Finally, we find that deprotonated PFAS molecules do not interact with MOF compounds and display a positive adsorption energy, with Bader charge calculations show a distinct difference between protonated and deprotonated PFAS molecules. Through this work, we disentangle how MOF and PFAS chemistry affects adsorption in this family of compounds.
期刊介绍:
The goal of Computational Materials Science is to report on results that provide new or unique insights into, or significantly expand our understanding of, the properties of materials or phenomena associated with their design, synthesis, processing, characterization, and utilization. To be relevant to the journal, the results should be applied or applicable to specific material systems that are discussed within the submission.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
