Identification and Validation of Cytotoxicity-Related Features to Predict Prognostic and Immunotherapy Response in Patients with Clear Cell Renal Cell Carcinoma.
{"title":"Identification and Validation of Cytotoxicity-Related Features to Predict Prognostic and Immunotherapy Response in Patients with Clear Cell Renal Cell Carcinoma.","authors":"Junxiao Yu, Bowen Zhao, You Yu","doi":"10.1155/2024/3468209","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Clear cell renal cell carcinoma (ccRCC) is a renal cortical malignancy with a complex pathogenesis. Identifying ideal biomarkers to establish more accurate promising prognostic models is crucial for the survival of kidney cancer patients.</p><p><strong>Methods: </strong>Seurat R package was used for single-cell RNA-sequencing (scRNA-seq) data filtering, dimensionality reduction, clustering, and differentially expressed genes analysis. Gene coexpression network analysis (WGCNA) was performed to identify the cytotoxicity-related module. The independent cytotoxicity-related risk model was established by the survival R package, and Kaplan-Meier (KM) survival analysis and timeROC with area under the curve (AUC) were employed to confirm the prognosis and effectiveness of the risk model. The risk and prognosis in patients suffering from ccRCC were predicted by establishing a nomogram. A comparison of the level of immune infiltration in different risk groups and subtypes using the CIBERSORT, MCP-counter, and TIMER methods, as well as assessment of drug sensitivity to conventional chemotherapeutic agents in risk groups using the pRRophetic package, was made.</p><p><strong>Results: </strong>Eleven ccRCC subpopulations were identified by single-cell sequencing data from the GSE224630 dataset. The identified cytotoxicity-related T-cell cluster and module genes defined three cytotoxicity-related molecular subtypes. Six key genes (SOWAHB, SLC16A12, IL20RB, SLC12A8, PLG, and HHLA2) affecting prognosis risk genes were selected for developing a risk model. A nomogram containing the RiskScore and stage revealed that the RiskScore contributed the most and exhibited excellent predicted performance for prognosis in the calibration plots and decision curve analysis (DCA). Notably, high-risk patients with ccRCC demonstrate a poorer prognosis with higher immune infiltration characteristics and TIDE scores, whereas low-risk patients are more likely to benefit from immunotherapy.</p><p><strong>Conclusions: </strong>A ccRCC survival prognostic model was produced based on the cytotoxicity-related signature, which had important clinical significance and may provide guidance for ccRCC treatment.</p>","PeriodicalId":12778,"journal":{"name":"Genetics research","volume":"2024 ","pages":"3468209"},"PeriodicalIF":2.1000,"publicationDate":"2024-08-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11379509/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genetics research","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1155/2024/3468209","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Clear cell renal cell carcinoma (ccRCC) is a renal cortical malignancy with a complex pathogenesis. Identifying ideal biomarkers to establish more accurate promising prognostic models is crucial for the survival of kidney cancer patients.
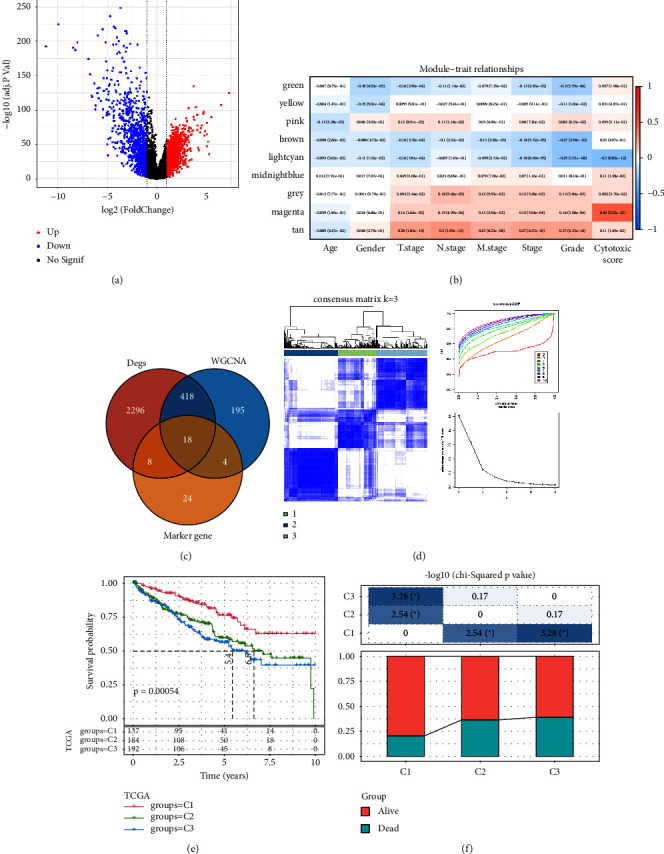
Methods: Seurat R package was used for single-cell RNA-sequencing (scRNA-seq) data filtering, dimensionality reduction, clustering, and differentially expressed genes analysis. Gene coexpression network analysis (WGCNA) was performed to identify the cytotoxicity-related module. The independent cytotoxicity-related risk model was established by the survival R package, and Kaplan-Meier (KM) survival analysis and timeROC with area under the curve (AUC) were employed to confirm the prognosis and effectiveness of the risk model. The risk and prognosis in patients suffering from ccRCC were predicted by establishing a nomogram. A comparison of the level of immune infiltration in different risk groups and subtypes using the CIBERSORT, MCP-counter, and TIMER methods, as well as assessment of drug sensitivity to conventional chemotherapeutic agents in risk groups using the pRRophetic package, was made.
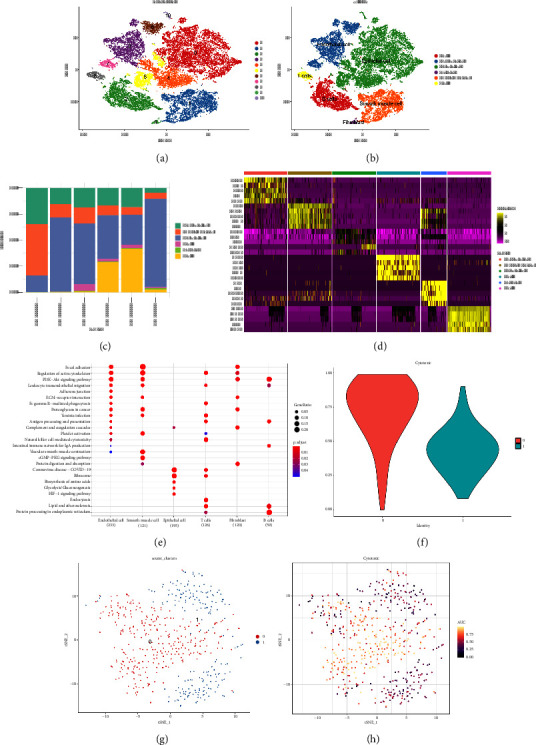
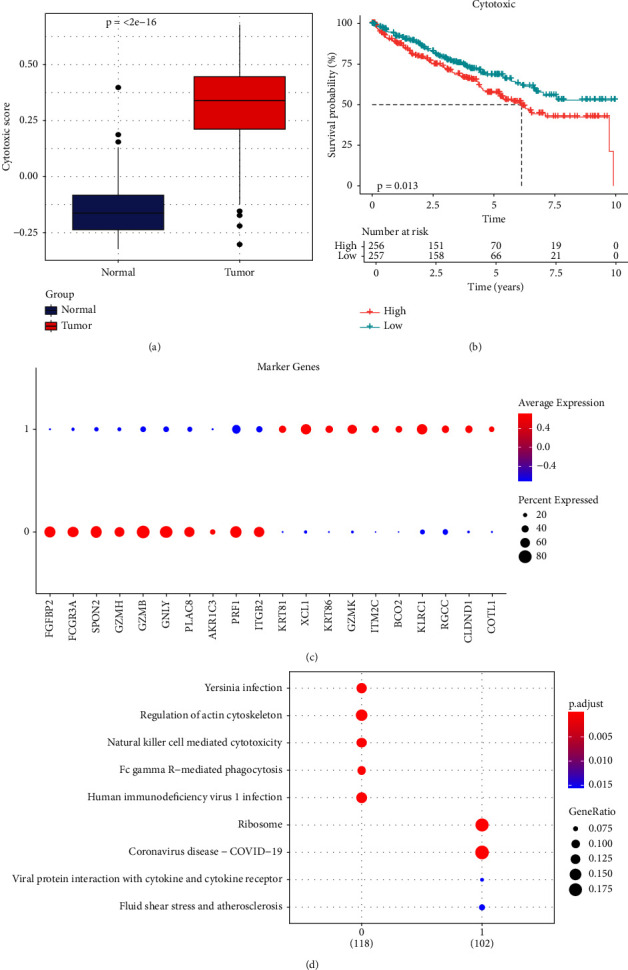
Results: Eleven ccRCC subpopulations were identified by single-cell sequencing data from the GSE224630 dataset. The identified cytotoxicity-related T-cell cluster and module genes defined three cytotoxicity-related molecular subtypes. Six key genes (SOWAHB, SLC16A12, IL20RB, SLC12A8, PLG, and HHLA2) affecting prognosis risk genes were selected for developing a risk model. A nomogram containing the RiskScore and stage revealed that the RiskScore contributed the most and exhibited excellent predicted performance for prognosis in the calibration plots and decision curve analysis (DCA). Notably, high-risk patients with ccRCC demonstrate a poorer prognosis with higher immune infiltration characteristics and TIDE scores, whereas low-risk patients are more likely to benefit from immunotherapy.
Conclusions: A ccRCC survival prognostic model was produced based on the cytotoxicity-related signature, which had important clinical significance and may provide guidance for ccRCC treatment.
期刊介绍:
Genetics Research is a key forum for original research on all aspects of human and animal genetics, reporting key findings on genomes, genes, mutations and molecular interactions, extending out to developmental, evolutionary, and population genetics as well as ethical, legal and social aspects. Our aim is to lead to a better understanding of genetic processes in health and disease. The journal focuses on the use of new technologies, such as next generation sequencing together with bioinformatics analysis, to produce increasingly detailed views of how genes function in tissues and how these genes perform, individually or collectively, in normal development and disease aetiology. The journal publishes original work, review articles, short papers, computational studies, and novel methods and techniques in research covering humans and well-established genetic organisms. Key subject areas include medical genetics, genomics, human evolutionary and population genetics, bioinformatics, genetics of complex traits, molecular and developmental genetics, Evo-Devo, quantitative and statistical genetics, behavioural genetics and environmental genetics. The breadth and quality of research make the journal an invaluable resource for medical geneticists, molecular biologists, bioinformaticians and researchers involved in genetic basis of diseases, evolutionary and developmental studies.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
