{"title":"Range Separation of the Interaction Potential in Intermolecular and Intramolecular Symmetry-Adapted Perturbation Theory","authors":"Du Luu, Clemence Corminboeuf, Konrad Patkowski","doi":"10.1021/acs.jctc.4c00608","DOIUrl":null,"url":null,"abstract":"Symmetry-adapted perturbation theory (SAPT) is a popular and versatile tool to compute and decompose noncovalent interaction energies between molecules. The intramolecular SAPT (ISAPT) variant provides a similar energy decomposition between two nonbonded fragments of the same molecule, covalently connected by a third fragment. In this work, we explore an alternative approach where the noncovalent interaction is singled out by a range separation of the Coulomb potential. We investigate two common splittings of the 1/r potential into long-range and short-range parts based on the Gaussian and error functions, and approximate either the entire intermolecular/interfragment interaction or only its attractive terms by the long-range contribution. These range separation schemes are tested for a number of intermolecular and intramolecular complexes. We find that the energy corrections from range-separated SAPT or ISAPT are in reasonable agreement with complete SAPT/ISAPT data. This result should be contrasted with the inability of the long-range multipole expansion to describe crucial short-range charge penetration and exchange effects; it shows that the long-range interaction potential does not just recover the asymptotic interaction energy but also provides a useful account of short-range terms. The best consistency is attained for the error-function separation applied to all interaction terms, both attractive and repulsive. This study is the first step toward a fragmentation-free decomposition of intramolecular nonbonded energy.","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"16 1","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2024-09-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00608","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
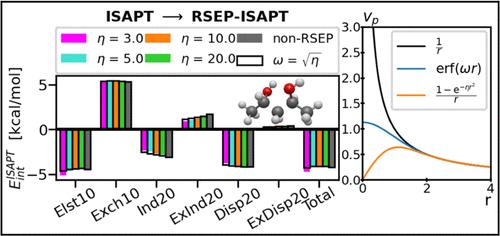
Symmetry-adapted perturbation theory (SAPT) is a popular and versatile tool to compute and decompose noncovalent interaction energies between molecules. The intramolecular SAPT (ISAPT) variant provides a similar energy decomposition between two nonbonded fragments of the same molecule, covalently connected by a third fragment. In this work, we explore an alternative approach where the noncovalent interaction is singled out by a range separation of the Coulomb potential. We investigate two common splittings of the 1/r potential into long-range and short-range parts based on the Gaussian and error functions, and approximate either the entire intermolecular/interfragment interaction or only its attractive terms by the long-range contribution. These range separation schemes are tested for a number of intermolecular and intramolecular complexes. We find that the energy corrections from range-separated SAPT or ISAPT are in reasonable agreement with complete SAPT/ISAPT data. This result should be contrasted with the inability of the long-range multipole expansion to describe crucial short-range charge penetration and exchange effects; it shows that the long-range interaction potential does not just recover the asymptotic interaction energy but also provides a useful account of short-range terms. The best consistency is attained for the error-function separation applied to all interaction terms, both attractive and repulsive. This study is the first step toward a fragmentation-free decomposition of intramolecular nonbonded energy.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
