{"title":"Pyrolytic conversion of glucose into hydroxymethylfurfural and furfural: Benchmark quantum-chemical calculations","authors":"Roberto López, Dimas Suárez","doi":"10.1002/jcc.27503","DOIUrl":null,"url":null,"abstract":"<p>Quantum chemical methods have been intensively applied to study the pyrolytic conversion of glucose into hydroxymethylfurfural (HMF) and furfural (FF). Herein, we collect the most relevant mechanistic proposals from the recent literature and organize them into a single reaction network. All the transition structures (TSs) and intermediates are characterized using highly accurate ab initio methods and the possible reaction pathways are assessed in terms of the Gibbs energies of the TSs and intermediates with respect to β-glucopyranose, selecting a 2D ideal-gas standard state at 773 K to represent the pyrolysis conditions. Several pathways can lead to the formation of both HMF and FF passing through rate-determining TSs that have <i>ΔG</i><sup>‡</sup> values of ~49–50 kcal/mol. Both water-assisted mechanisms and nonspecific environmental effects have a minor impact on the Gibbs energy profiles. We find that the HMF → FF + CH<sub>2</sub>O fragmentation has a small <i>Δ</i><sub>rxn</sub><i>G</i> value and an accessible <i>ΔG</i><sup>‡</sup> barrier. Our computational results, which are in consonance with the kinetic parameters derived from lumped models, the results of isotopic labeling experiments and the reported HMF/FF molecular ratios, could be useful for modeling studies including on nonequilibrium kinetic effects that may render more information about product yields and the relevance of the various pathways.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 32","pages":"2991-3003"},"PeriodicalIF":4.8000,"publicationDate":"2024-09-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27503","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27503","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
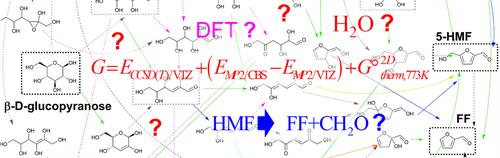
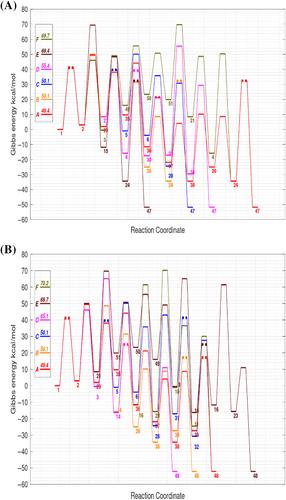
Quantum chemical methods have been intensively applied to study the pyrolytic conversion of glucose into hydroxymethylfurfural (HMF) and furfural (FF). Herein, we collect the most relevant mechanistic proposals from the recent literature and organize them into a single reaction network. All the transition structures (TSs) and intermediates are characterized using highly accurate ab initio methods and the possible reaction pathways are assessed in terms of the Gibbs energies of the TSs and intermediates with respect to β-glucopyranose, selecting a 2D ideal-gas standard state at 773 K to represent the pyrolysis conditions. Several pathways can lead to the formation of both HMF and FF passing through rate-determining TSs that have ΔG‡ values of ~49–50 kcal/mol. Both water-assisted mechanisms and nonspecific environmental effects have a minor impact on the Gibbs energy profiles. We find that the HMF → FF + CH2O fragmentation has a small ΔrxnG value and an accessible ΔG‡ barrier. Our computational results, which are in consonance with the kinetic parameters derived from lumped models, the results of isotopic labeling experiments and the reported HMF/FF molecular ratios, could be useful for modeling studies including on nonequilibrium kinetic effects that may render more information about product yields and the relevance of the various pathways.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
