{"title":"MMFA-DTA: Multimodal Feature Attention Fusion Network for Drug-Target Affinity Prediction for Drug Repurposing Against SARS-CoV-2","authors":"Guanxing Chen, Haohuai He, Qiujie Lv, Lu Zhao, Calvin Yu-Chian Chen","doi":"10.1021/acs.jctc.4c00663","DOIUrl":null,"url":null,"abstract":"The continuous emergence of novel infectious diseases poses a significant threat to global public health security, necessitating the development of small-molecule inhibitors that directly target pathogens. The RNA-dependent RNA polymerase (RdRp) and main protease (Mpro) of SARS-CoV-2 have been validated as potential key antiviral drug targets for the treatment of COVID-19. However, the conventional new drug R&D cycle takes 10–15 years, failing to meet the urgent needs during epidemics. Here, we propose a general multimodal deep learning framework for drug repurposing, MMFA-DTA, to enable rapid virtual screening of known drugs and significantly improve discovery efficiency. By extracting graph topological and sequence features from both small molecules and proteins, we design attention mechanisms to achieve dynamic fusion across modalities. Results demonstrate the superior performance of MMFA-DTA in drug-target affinity prediction over several state-of-the-art baseline methods on Davis and KIBA data sets, validating the benefits of heterogeneous information integration for representation learning and interaction modeling. Further fine-tuning on COVID-19-relevant bioactivity data enhances model predictions for critical SARS-CoV-2 enzymes. Case studies screening the FDA-approved drug library successfully identify etacrynic acid as the potential lead compound against both RdRp and Mpro. Molecular dynamics simulations further confirm the stability and binding affinity of etacrynic acid to these targets. This study proves the great potential and advantages of deep learning and drug repurposing strategies in supporting antiviral drug discovery. The proposed general and rapid response computational framework holds significance for preparedness against future public health events.","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"43 1","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2024-09-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00663","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
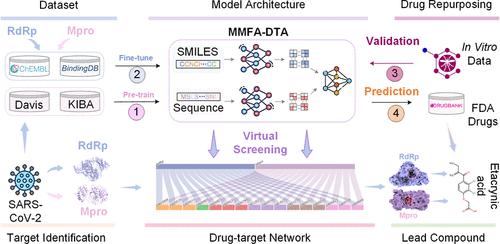
Abstract
The continuous emergence of novel infectious diseases poses a significant threat to global public health security, necessitating the development of small-molecule inhibitors that directly target pathogens. The RNA-dependent RNA polymerase (RdRp) and main protease (Mpro) of SARS-CoV-2 have been validated as potential key antiviral drug targets for the treatment of COVID-19. However, the conventional new drug R&D cycle takes 10–15 years, failing to meet the urgent needs during epidemics. Here, we propose a general multimodal deep learning framework for drug repurposing, MMFA-DTA, to enable rapid virtual screening of known drugs and significantly improve discovery efficiency. By extracting graph topological and sequence features from both small molecules and proteins, we design attention mechanisms to achieve dynamic fusion across modalities. Results demonstrate the superior performance of MMFA-DTA in drug-target affinity prediction over several state-of-the-art baseline methods on Davis and KIBA data sets, validating the benefits of heterogeneous information integration for representation learning and interaction modeling. Further fine-tuning on COVID-19-relevant bioactivity data enhances model predictions for critical SARS-CoV-2 enzymes. Case studies screening the FDA-approved drug library successfully identify etacrynic acid as the potential lead compound against both RdRp and Mpro. Molecular dynamics simulations further confirm the stability and binding affinity of etacrynic acid to these targets. This study proves the great potential and advantages of deep learning and drug repurposing strategies in supporting antiviral drug discovery. The proposed general and rapid response computational framework holds significance for preparedness against future public health events.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
