Pablo A. Denis, Jose A. S. Laranjeira, Nicolas F. Martins, Julio R. Sambrano
{"title":"Codoped germanene with 3p and 4p elements elements","authors":"Pablo A. Denis, Jose A. S. Laranjeira, Nicolas F. Martins, Julio R. Sambrano","doi":"10.1007/s00894-024-06133-6","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>The relentless need for new materials to be used in electronic devices has opened new research directions in materials science. One of them involves using two-dimensional materials, among which there is current interest in using germanene. The heteroatom doping of germanene has been proposed as a possible approach to fine-tuning its electronic properties. However, this procedure is complicated because locating the dopants with a specific arrangement is challenging, thus achieving reproducibility. To avoid this problem, we propose the codoping of germanene to understand if dopants prefer to be agglomerated as observed for graphene or if they prefer to adopt a random disposition. Herein, we employed first-principles calculations to study 21 codoped germanene systems with one <i>3p</i> (Al, Si, P, and S) and one <i>4p</i> (Ga, As, and Se) element. Our results indicate that in the cases of AlP, AlS, GaP, GaS, GaAs, and GaSe codoped germanene, the dopants show a tendency to be located in specific lattice positions. The ortho disposition of dopants is preferred for AlP, AlS, GaP and GaS codoped germanene and their <i>4p</i> counterparts GaAs and GaSe codoped germanene, and the materials showed interesting electronic properties making them suitable to develop germanene-based electronic materials.</p><h3>Methods</h3><p>We utilized the M06-L, HSE06 methods accompanied by the 6-31G* basis sets to perform periodic boundary conditions calculations as implemented in Gaussian 09. The unit cells were sampled employing 100 k-points for geometry optimizations and 2000 k-points for electronic properties The ultrafine grid was employed. Results were visualized employing Gaussview 5.0.1. In addition to this, we performed B3LYP-D3 periodic calculations as implemented in CRYSTAL17.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"30 10","pages":""},"PeriodicalIF":2.1000,"publicationDate":"2024-09-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-024-06133-6","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Context
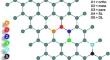
The relentless need for new materials to be used in electronic devices has opened new research directions in materials science. One of them involves using two-dimensional materials, among which there is current interest in using germanene. The heteroatom doping of germanene has been proposed as a possible approach to fine-tuning its electronic properties. However, this procedure is complicated because locating the dopants with a specific arrangement is challenging, thus achieving reproducibility. To avoid this problem, we propose the codoping of germanene to understand if dopants prefer to be agglomerated as observed for graphene or if they prefer to adopt a random disposition. Herein, we employed first-principles calculations to study 21 codoped germanene systems with one 3p (Al, Si, P, and S) and one 4p (Ga, As, and Se) element. Our results indicate that in the cases of AlP, AlS, GaP, GaS, GaAs, and GaSe codoped germanene, the dopants show a tendency to be located in specific lattice positions. The ortho disposition of dopants is preferred for AlP, AlS, GaP and GaS codoped germanene and their 4p counterparts GaAs and GaSe codoped germanene, and the materials showed interesting electronic properties making them suitable to develop germanene-based electronic materials.
Methods
We utilized the M06-L, HSE06 methods accompanied by the 6-31G* basis sets to perform periodic boundary conditions calculations as implemented in Gaussian 09. The unit cells were sampled employing 100 k-points for geometry optimizations and 2000 k-points for electronic properties The ultrafine grid was employed. Results were visualized employing Gaussview 5.0.1. In addition to this, we performed B3LYP-D3 periodic calculations as implemented in CRYSTAL17.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
